
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Review Article - Open Access, Volume 4
Studies on the mutagenic and tumor-promoting effects of TRF2 protein in the mechanism of tumor formation
Zhengyi Wang1*; Liang Zhou2; Xiaoying Wu3
1GCP center, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, Chengdu city, Sichuan province, China. Institute of Laboratory Animals of Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, CHENGDU, SICHUAN, CN. Yinglong Community, Zhonghe Street, High-tech Zone, Chengdu City, Sichuan Province, China.
2Institute of Laboratory Animals of Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, CHENGDU, SICHUAN, CN.Yinglong Community, Zhonghe Street, High-tech Zone, Chengdu City, Sichuan Province, China.
3Ministry of education and training, CHENGDU second people’s hospital, chengdu city, Sichuan province, China.
*Corresponding Author : Zhengyi Wang
GCP center, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, chengdu city, sichuan province, China; Institute of Laboratory Animals of Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, chengdu city, sichuan province, China;
Email: 983927965@qq.com
Received : May 04, 2023
Accepted : Jun 09, 2023
Published : Jun 16, 2023
Archived : www.jcimcr.org
Copyright : © Wang Z (2023).
Abstract
In recent years, the gene mutation hypothesis related to tumor formation has been deeply studied, and some scholars have put forward some new views on the basis of the original theory. The view that tumor formation begins with the genetic material variation of somatic cells has been confirmed and universally recognized by the academic community. But what is the relationship between macroscopic chromosomal aberrations and microscopic gene mutations in the process of tumor formation? Does the two interact synergistically to promote tumor formation, or does it require only one of them to complete tumor transformation, or does it require other cofactors to work together? Based on the progress of tumor research in recent years, the author of this article focuses on the two stages of tumor formation: somatic mutagenesis stage and tumor promotion stage, and discusses the related factors that affect somatic cell mitosis, the process of aneuploidy formation, and the transformation of tumor microenvironment by tumor promoting factors. Among them, Telomere Repeat Sequence Binding Protein 2 (TRF2) plays a very key role in the process of tumor formation: it is not only an important protein that leads to the imbalance of somatic mitosis and the continuous instability of chromosome, but also an important factor to maintain the relative stability of chromosome of immortalized tumor cells and promote the mitosis of tumor cells; At the same time, it has a major impact on the stability of tumor suppressor genes or oncogenes. In addition, it is a cellular environmental tumor-promoting factor that regulates tumor cell microenvironment and promotes tissue colonization and rapid proliferation of tumor-initiating cells. The expression level of TRF2 in different periods of cells caters to the needs of different periods of tumor formation, determines the direction of tumor formation and development, and is an important direction for in-depth research on tumor prevention and clinical treatment in the future.
Keywords: TRF2; Tumor formation; Tumor mutagenesis; Environmental promoting effect.
Citation: Wang Z, Zhou L, Wu X. Studies on the mutagenic and tumor-promoting effects of TRF2 protein in the mechanism of tumor formation. J Clin Images Med Case Rep. 2023; 4(6): 2459.
Introduction
The study of tumor formation mechanism has always been the core problem of modern medicine trying to break through [1]. The current view is that the root cause of tumor formation is genetic mutation [2-4]. Most research has focused on oncogenic susceptibility to genetic mutations and their treatment options. In terms of cladistics, human tumors can be divided into three types: hereditary familial tumor syndrome (monogenic inheritance), polygenic tumor genetic susceptibility and local tumor cell genetic changes (acquired). It has long been believed that tumor-driven genes play a decisive role in the development of cancer. However, recent studies have found a large number of somatic genetic mutations in a number of normal adult tissues, including skin, esophagus, colon, liver, lung, and endometrium, suggesting that DNA mutations are common in healthy people [5-12]. The mutated cells that did not cause a disease response also included mutations in certain tumor-driving genes. For example, 30% to 80% of cells in normal esophageal tissue of the elderly contain mutations in the oncogenic driver gene NOTCH1 [13,6]. However, the mutated cells did not form tumor tissue and remained in healthy people for a long time. Rodent carcinogenicity test studies have shown that the main risk factor for cancer in mice is not persistent strong carcinogenic mutations in somatic genes [14,15]. Such cells carrying strong oncogene mutations can persist in healthy individuals for long periods of time without tumor transformation. Tumor can only be induced under the action of environmental tumor promoting mediators (tumor promoters), that is, there are some more direct tumor promoting factors in the process of tumor formation [14,15]. Based on the above facts, it is proved that the simple mutation of cancer driver gene or tumor suppressor gene does not determine the formation of tumor structure, so there should be some more direct induction component in the body for the transformation and development of human tumors, and what factors play a decisive role?
Some questions about the mutation hypothesis in cancer genes
It has been shown that the formation of human tumors is closely related to abnormal hereditary material in cells [11,16,17]. There are many hypotheses about the hereditary mechanism of tumor formation, such as chromosome imbalance hypothesis [18], double mutation hypothesis [19], transformed gene hypothesis [20], on cogene hypothesis [21,22], etc. Based on the observation that the mutation load of tumor cells exceeds that of normal aging tissue cells, some scholars believe that although there are many mutations in healthy individuals, the reason why malignant progression is relatively rare may be that the number of mutations or combinations of mutations is below the minimum threshold level required for complete transformation of the tumor [8,13]. However, this view does not account for the fact that tumor tissue is not generated in mice under long-term induction of strong on cogenic mutations that lead to cancer driver gene mutations beyond the threshold [23,24]. Conversely, treatment of these mutant cells with environmental tumor promoters (which no longer induce the accumulation of gene mutations) induces tumor formation in a short time [23,24]. These mutated cells resemble dormant tumor cells in the human body and require appropriate environmental changes to ensure tumor formation [25,26]. ThereTherefore, it is speculated that on cogene mutations are only a factor or initial step in tumor formation, and there must be another tumor rate-limiting mechanism that plays a decisive role in regulation. The controllability of this rate-limiting mechanism will determine the prognosis of tumor development. It is generally believed that the regulation of the internal and external microenvironment of mutant cells plays a key role in the rate-limiting mechanism of tumor progression.
In conclusion, during the transformation from normal cells to tumor cells, mutations of tumor driver genes with a high threshold may lead to the formation of mutant cells (cancer cells), but cannot lead to the occurrence of malignant tumors. Research on the rate-limiting mechanism of malignant tumor formation seems to have more clinical value. This rate-limiting mechanism is closely related to the changes in the peripheral microenvironment of mutant cells, and the changes in the levels of some proteins in mutant cells may be the initiating factors of the changes in the peripheral microenvironment. The study of these protein molecules is more conducive to the inhibition of local colonization of mutant cells and the prevention of tumor occurrence.
Molecular mechanisms underlying the aneuploidy hypothesis in tumor formation
A large number of studies have found that the chromosomes of solid tumor cells are all aneuploid [27-29]. This particular change in cell chromosomal structure seems to be the only way ultimately to form tumor tissue [27-31]. Based on these facts, some scholars have proposed the hypothesis of aneuploidy in tumorigenesis [32,33]. Exposure to carcinogenic factors, such as physical or chemical, can lead to cell chromosome damage and deformation, and may lead to aneuploidy. Aneuploidy may result in abnormal expression of mitoid-related genes, abnormal spindle protein ratio, and even abnormal centriole number [34,35]. Unbalanced spindles can lead to random loss or acquisition of certain chromosomes [33]. During each cell division, these aneuploidy chromosomes are randomly grouped and asymmetrically distributed into descendant cells [33]. This vicious cycle promotes the aneuploidy itself to catalyze the karyotype change and evolution of cells, ultimately leading to the karyotype of tumor cells [33]. The hypothesis holds that the occurrence of tumor is the result of aneuploid autocatalysis and is not directly related to gene mutation. However, the hypothesis does not well explain the rate-limiting mechanism of dormant tumor cells, and it is still insufficient to explain the development and outcome of tumor cells in human body and guide clinical tumor treatment.
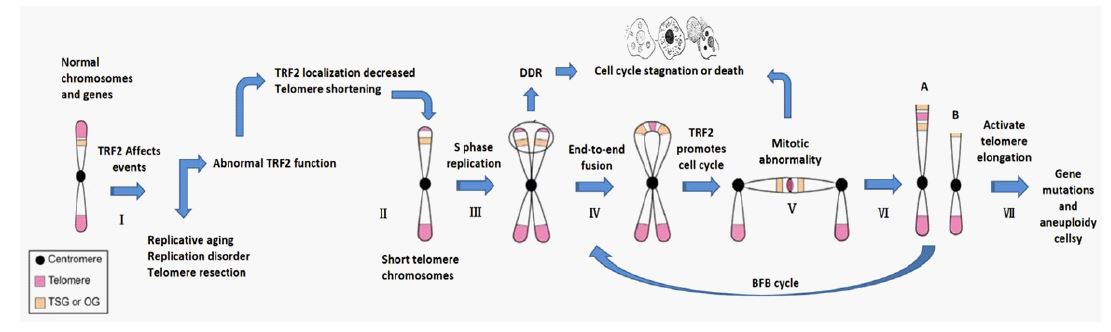
According to the aneuploid theory of tumor formation, various chromosomal aneuploid aberrations are potentially carcinogenic, such as ionizing radiation, ultraviolet radiation, chemical substances, etc. TRF2 often plays a very important role in these induction mechanisms. For example: Replicative senescence is associated with severe telomere shortening and decreased TRF2, or catastrophic telomere removal due to TRF2 dysfunction, and abnormal DNA removal induced by various heterochromatin replication disorders associated with TRF2 [36-38]. Among these factors, TRF2 localization in the DNA repeat structure is lost, thus removing the protection of telomere and heterochromatin sequences. Exposed telomere ends or heterochromatin broken ends are identified by cells as DNA Double-Strand Breaks (DSB), which activate the cell cycle regulator protein-ATM (Ataxia telangiectasia mutant kinase), block cell cycle progression, and induce DNA Damage Repair (DDR) cascades [39]. In the subsequent process, on the one hand, if DSB foci cannot connect with other chromosomal ends (or DNA broken ends) in time, the cell cycle continues to be blocked, and the related proteins in the cascade reaction can induce and start the apoptosis process to remove the functional defective cells and avoid cell mutation. At this time, if the suppressive protein genes related to apoptosis are mutated (such as ATM,TP53, etc.), cells can bypass the prevention and control mechanism of apoptosis and make defective cells enter the mitosis cycle, resulting in Chromosome Instability (CIN) during division and abnormal amplification of the mutant cells. On the other hand, if DSB foci can connect with other chromosomal ends (DNA broken ends) in time, DSB foci can be repaired abnormally, and ATM-related cascade proteins can be eliminated, releasing cell cycle arrest and entering mitotic crisis [40-42]. Some studies have found that during the crisis, loss of TRF2 can lead to cessation of mitosis, and most cells die due to detachment from the cell cycle, thus preventing malignant transformation [40,41,43]. However, individual cells can maintain a continuous chromosome recombination or Breaking-Fusion-Bridge (BFB) cycle and continue mitosis, resulting in extremely unstable chromosomes, amplification of a large number of aneuploid cells, and self-catalytic karyotype mutation (or evolution), resulting in tumor karyotypes [44]. Due to aneuploid recombination, the frequency of some growth-related genes changes, and cells can acquire abnormal abilities after the expression of certain genes [45]. For example, increasing the expression level of hTERT gene can activate the long-term survival mechanism of cells. The telomere is lengthened by telomerase, which ensures the location of TRF2 in repeat sequence and maintains telomere and heterochromatin structure. It can prevent the malignant cycle of chromosome abnormal cells from recombination, improve the chromosome instability of mutant cells, and maintain their survival ability. The mutated cells can survive permanently and eventually form tumor-initiating cells [44,45]. This process may be related to the action mechanism of various exogenous teratogenic factors at cellular and molecular levels. TRF2 maintains the structural and functional state of heterochromatin such as telomeres and plays an important role in maintaining chromosome stability [46]. Many factors contribute to abnormal changes in the level or function of TRF2 in the nucleus, and too high or too low TRF2 levels can damage chromosomes, lead to instability, and induce cellular tumor transformation [47-50]. In fact, clinical studies have found that older adults associated with TRF2 abnormalities or chromosomal aberrations such as Bloom syndrome and Werner syndrome are closely associated with a high incidence of tumors [51,52]. In conclusion, TRF2 may be a key protein in the formation of aneuploid cells, and its abnormal function is closely related to tumorigenesis.
The aneuploidy hypothesis seems to offer a plausible explanation for tumor cell evolution in many ways. However, the transformation from somatic cells to primary tumor cells does not imply the formation of tumor bodies. For example, Circulating tumor cells in asymptomatic patients - Circulating Tumor Cells (CTC) -- are dormant in the circulating system in most cases because there is no suitable peripheral transplant environment. They do not easily colonize and form tumors. They require awakening by environmental tumor promoter [53-55]. Similarly, progression from tumor initiating cells to tumors requires appropriate environmental tumor promoters and sufficient time to transform the Tumor Cell Peripheral Microenvironment (TME). So that the tumor can survive, get rid of the pressure of being eliminated, and provide guarantee for its rapid reproduction and invasion. The tumor immunosurveillance hypothesis explains the non-inherited determinants of tumorigenesis [56]. The incidence of tumor diseases is much higher in people with weakened immune systems such as chronic wasting diseases, AIDS, organ transplantation or aging [57,58]. It has been proved that the human immune system can recognize and destroy the initiating cells of neoplasm, indicating the existence of specific immune antigens in neoplasm cells. The ability of the body’s immune system to recognize these antigens supports the hypothesis of immune surveillance [56]. Immunosurveillance is an essential part of the cancer immunoediting process [59-62]. Under the action of immune pressure, immune editing has the dual role of preventing host cancer and evading tumor immunity [63]. Compared with normal cells, the presence of tumor-specific antigen proteins indicated that tumor cells did synthesize a variety of new tumor-specific proteins due to some gene mutations. It has been proved that the mutation load of tumor cells determines the prognosis of immunotherapy [64,65]. Since the classical aneuploid hypothesis of tumorigenesis does not provide a clear explanation of the role of gene mutations in tumorigenesis, changes in gene frequency (amplification or deletion) caused by chromosome recombination alone cannot explain the molecular mechanism of the origin of multiple specific tumor proteins. Based on the aneuploidy hypothesis and recent studies on telomere related proteins in tumor cells, we speculate that the heterochromatin localization and functional status of TRF2 protein play a decisive role in the transformation of normal cells into tumor cells. First, the local localization of TRF2 decreased or dysfunction, resulting in chromosome damage. Under the action of DNA damage repair mechanism, the surviving cells can undergo chromosome end-to-end fusion, increase the number of centromeres or abnormal spindle structure. Driven by cyclins, mitosis is disordered or delayed, while irregular or random chromosome division occurs. The BFB cycle repeats itself over and over again, leading to massive DNA damage and recombinant mutations. The surviving cells underwent a telomere crisis, and large-scale recombination mutations occurred in DNA at the same time as the aneuploid structure was formed. Survival cells activate telomere elongation mechanisms during chromosome recombination, such as increased telomerase expression or telomere homologous recombination, to produce extended telomere repeats at the ends of mutant cells. As TRF2 relocates and plays a protective role in repeated sequences, the surviving cells gradually restore basic telomere function, the end-to-end fusion cycle terminates, and the mutant cells enter a relatively stable division cycle. Due to early large-scale recombination, the surviving cells at this time are not normal cells but aneuploid cells with a large number of mutated genes or specific tumor proteins. Here we only speculate about the possible relationship between tumor aneuploid modification and gene mutation, and the actual situation needs further study.
In conclusion, the formation of initial tumor cells may be a synergistic process of simultaneous pathological changes of gene mutation and chromosome mutation (aneuploid). First of all, CIN caused by physical, chemical and other extracellular inducements, or directly due to gene mutations of endogenous important proteins that can induce CIN, can lead to the generation of initial tumor cells. Secondly, large - scale gene mutations will occur in the process of chromosomal aberration modifica tion due to CIN. CIN is aggravated by mutations in genes that affect cell proliferation, DNA repair, and proteins in telomeres or heterochromatin regions of chromosomes [66]. Thus, the formation of tumor-initiating cells is a combination of microscopic genetic mutations and macroscopic chromosomal aberrations. It can be caused by changes in gene function, such as HRAS, MYC, TP53, TRF2, Rb1, CHEK2 and other cancer-related genes (including gene mutations, frequency changes, epigenetics, biochemical pathways and adjacent network signal transduction effects) [67-72]. These changes in gene function affect homeostasis of a range of key cellular functions. Examples include the mitotic cycle, heterochromatin function, or chromosome stability [73]. It can also directly cause chromosomal damage by physical and chemical factors of external environmental conditions, especially heterochromatin dysfunction, but ultimately cannot avoid the fate of tumor cell gene mutation and chromosome aneuploidy (Figure 1).
Relationship between tumor-forming mechanism and TRF2 in telomere Shelterin complex
Shelterin protein complex is an essential component for the maintenance of intact telomere structure. In all mammals, Telomere Consists Of A Highly Conserved Hemeric (TTAGGG) tandem repeat DNA sequence and a telomere binding protein, which in turn consists of a Shelterin complex, accessory factors, and telomerase. The human Shelterin complex consists of six core proteins: Telomeric Rest-Binding Factors 1 and 2 (TRF1, TRF2), TERF1-Interacting Protein 2 (TIN2), Repressor Activator Protein 1 (RAP1), Protection of telomeres 1(POT1) and Telomere-binding protein pot1-branched protein 1(TPP1). TRF2 is an important polypeptide in telomere Shelterin complex. The gene is located on chromosome 16Q22.1 with a length of 30 Kb and is widely expressed in human body. TRF2 protein in shelterin complex is a telomere protective protein, which plays an important role in maintaining the normal function of telomeres. For example, TRF2 folds human telomeres into loops to prevent unwanted DDR, blocks ATM signals and NHEJ (Non-Homologous End Junctions), promotes telomere replication, and is a key regulator of telomere integrity. Tumorigenesis is related to chromosome instability, and the incomplete structure and function of telomere is an important factor causing chromosome instability. Based on the decisive role of TRF2 in telomere structure and function, the relationship between TRF2 and different stages of tumor genesis is discussed below
The role of TRF2 in the transformation of somatic cells into tumor cells
The transformation of a somatic cell into a tumor cell involves unstable changes in previously stable hereditary material in the nucleus. It is an early stage of cancer transformation, culminating in the formation of tumor-initiating cells with stem-cell characteristics, and is therefore known as the tumor mutation stage or tumor initiation stage [74]. The transformation of somatic cells into tumor cells has solved the problem of tumor origin [75-77]. In the stage of tumor mutagenesis, induced by a variety of nucleic acid sensitive events inside and outside the body, such as replicative senescence, cell fusion events, ionizing radiation or chemical mutagenesis, can directly lead to the continuous accumulation of chromosome aberrations and gene mutations in normal cells [78-81]. At this stage, TRF2 protein has two opposite effects, which is of great significance for the formation and outcome of tumor-initiating cells.
Under physiological conditions, TRF2 can inhibit the transformation of somatic cells into tumor cells
The transformation of tumor cells results from chromosome instability. Stable chromosomes must have complete telomere functional structure, and its maintenance depends on two aspects: a. Dependent on the activity of telomerase or Alternative Lengthening Of Telomere (ALT), which compensates for replication erosion; b. Depending on its own special chromatin organization, it protects the ends of chromosomes from abnormal signals and repair. Chromatin mediated telomere protection involves several pathways regulated by proteins that directly or indirectly bind telomere DNA, such as the Shelter in complex [82,83]. TRF2 is a very important telomere protective protein in the Shelter in complex, which plays an important role in maintaining normal telomeres, stabilizing chromosomes and preventing transformation into tumor cells in the following three aspects.
ⅰ. TRF2 constructs and maintains the loop structure at the end of chromo somes, hides the telomere tails and stabilizes chromosomes. The TRF2 dimer wraps about 90 bp of DNA through several lysine and arginine residues located around its TRFH domain, forming a DNA topology and mediating the formation of loop structure [84-85]. It causes the repeated double-stranded DNA in the telomere to form a noose telomere loop (T-loop), and the 3’ end of the single strand in the telomere can be inserted into the Double-Stranded DNA (ds DNA) of the telomere (the 3’ protrusion is paired with the C-rich strand), replacing the g-rich strand in the double strand to form a displacement loop, also known as the D-loop. The loop structure of the telomere concealed the end of the chromosome and did not produce the DSB senter-like MRN complex, which interacts with the end of the DNA, thus preventing MRN-dependent ATM activation, eliminating the damage repair of the DSB at the end of the double-stranded DNA and keeping the telomere DNA stable, thus avoiding DDR or end-to-end fusion [86,84].
ⅱ. TRF2 can inhibit direct telomere DDR production and abnormal repair. In addition to inhibiting DDR by maintaining the telomere loop structure, TRF2 also regulates the activity of ATM cascade downstream proteins through its “Inhibitory DDR Response Domain” (IDDR). At γ-H2AX-labeled telomeres, TRF2 blocks the recruitment and inhibition of E3 ubiquitin ligase RNF168 in a dependent manner of deubiquitination enzyme BRCC3 and ubiquitin ligase UBR5 through the “IDDR motif” in its hinge domain. At the level of RNF168, the DNA damage signaling cascade is cut off and 53BP1 localization is prevented, thus preventing end-to-end chromosome fusion [87]. TRF2 is also a major inhibitor of the three basic modes of Telomere End-To-End Fusion (C-NHEJ, A-NHEJ, and HDR). It inhibits the repair of DNA abnormalities and reduces cell variation through negative regulation of Ku protein and inhibition of PARP1 activation [88-89].
ⅲ. TRF2 is an important protein to maintain the physiological length of telomeres. Telomere length is closely related to chromosome stability, and TRF2 is a key participant in telomere protection and telomere length maintenance, both preventing severe shortening caused by catastrophic telomere loop cutting and being a negative regulator of telomere length [90][91]. On the one hand, TRF2 prevents telomeres from becoming too short. TRF2 prevents catastrophic cleavage of telomeres through the interaction between its N-terminal basic domain and the ionase-related protein SLX4 and other different endonuclease types involved in the dissociation activities (e.g. GEN1, MUS81, etc.). At the same time, various types of enzymatic activity were examined through basic domains to promote precise repair of the stasis forks that occur during telomere replication. SLX4 is also recruited by the TRF2 homo-dimer domain. SLX4 is also a structure-specific DNA repair nuclease scaffold, which can attract the holliday-linked processing enzyme SLX1 and other nucleases to telomeres, prevent telomere damage, elongation and brittleness, and play a variety of roles in regulating telomere homeostatic [92]. On the other hand, TRF2 can promote telomere shortening by XPF-ERCC1 enzyme [93]. The TRF2-mediated Heregulin β2 (HRGβ2) growth factor of the Heregulin protein family is also a negative regulator of telomere length [94-95]. TRF2 can also regulate the expression of hTRET, the rate-limiting catalytic protein component of telomerase, to maintain telomere at appropriate lengths through “Telomere position effect -- over long distances (TPE-OLD)” effect [96]. Thus, the presence of TRF2 ensures the normal length of telomeres and is an important protein to prevent somatic aging and mutation.
Under pathological conditions, TRF2 can promote the transformation of somatic cells into tumor cells and stabilize the mutant cells
When under the influence of senescence or external mutagenic factors, the chroma of somatic cells is unstable. Some observations have found dynamic changes in intracellular TRF2 content and distribution (increasing or decreasing in different regions of different transformed cells), or changes or no changes in TRF2 mRNA transcription levels [87,88]. TRF2 is redistributed in the nucleus, usually with reduced distribution and loss of function in the telomere region. Telomere without the above-mentioned protective effect of TRF2 will activate ATM and ATR kinases, induce DDR cascade reaction, and cause cell proliferation cycle arrest, apoptosis or abnormal damage repair. Under the promotion of cyclin by TRF2, some cells bypassing the apoptosis pathway enter the mitotic crisis stage, passing through the BFB cycle, leading to irregular chromosome recombination, catalyzing the evolution of karyo type, and forming a large number of aneuploid progeny cells [97-101]. Some studies have found that knockout of TRF2 protein in cells can lead to mitotic arrest and cell death during cell crisis, thus preventing the occurrence of tumor diseases. This strongly suggests that TRF2 plays an indispensable role in promoting tumor formation [102,103]. The telomeres of recombinant chromosomes in progenitor cells still lack the protective effect of TRF2, and aneuploid tumor karyotypes are formed through repeated crisis cycles, large-scale fusion and recombination. Until the telomere production mechanism at the end of the chromosome of the recombinant cell is activated (e.g. Due to gene on chromosome fracture fragments after repeatedly superposition of restructuring, frequency of hTERT gene expression was promoted) or telomere repeat sequences at the end of the chromosome replacement, at this point, the chromosomes have enough length of telomere repeat sequences, some TRF2 specificity to combine again to repeat sequences, play the function of telomere protection, the telomere structure was maintained after cell recombination, thus blocking the BFB crisis cycle, enabling tumor karyotype cells to survive after chromosome recombination, and eventually forming initial tumor cells. However, unlike normal cells, the telomere function of TRF2 in the initial tumor cells was always at a low level, which may be related to the ability of KIP protein to regulate the binding of TRF2 to telomere [104].
Although the formation mechanism of initial tumor cells has been gradually understood, there is currently no effective reversal and recovery measures for chromosome recombination or gene mutation in clinical treatment strategies, and clinical intervention in the formation stage of tumor cells is still difficult to achieve [105]. Since this process involves changes in the content and distribution of the key protein TRF2 in cells, with the improvement of the research technology and functional understanding of telomere TRF2 protein, it seems possible to analyze the changes of TRF2 in cells to gain insight into the process that drives cell mutation transformation. And may provide strategies for early intervention targeting “tumour initiating cells”. It has been found that TRF2 levels are regulated in a cycle-dependent manner: during the interphase of cell division, TRF2 levels are low and appear to be limited to telomeres [99]. However, TRF2 levels increase in the S phase in a manner dependent on transcriptional regulation, and are up regulated with cycle changes. This is associated not only with increased levels of TRF2, but also with extra-telomere localization of TRF2, which is concentrated around chromosomes in mitotic cells, suggesting that TRF2 may have other important extra-telomere functions [99]. During the generation of tumor-initiating cells, the content of TRF2 in cells will also change or be redistributed: Generally, when cells are affected by mutagenesis factors such as radiation, the overall level of TRF2 will be up regulated, which may be because TRF2 recruits proteins to telomere and chromosome sequences to promote DNA repair. When cells are damaged and DDR repair occurs, telomere TRF2 levels are usually down-regulated to ensure terminal fusion when chromosomal end-to-end fusion occurs. Before the tumor-initiation cells acquired immortalization ability, the intracellular TRF2 level did not change significantly, but the TRF2 level at the end of chromosomes remained low, and the telomere lost the protection of TRF2, which enabled the BFB cycle to continue. After immortalization, intracellular TRF2 level may increase significantly (although TRF2 mRNA level does not change, the mechanism remains to be elucidated), which may be related to the accumulation of TRF2 in cells due to the reduction of TRF2 degradation protease action. Elevated TRF2 is dispersed in the nucleus, not only in the telomere region but also in other non-telomere regions, further suggesting that not all TRF2 is associated with telomeres in these cells and also regulates other functions of tumor-initiating cells [99-101]. The scattered distribution of TRF2 may be evidence of interactions between various proteins in the cell and TRF2 at different targets, which may regulate various signal transduction and protein generation in the cell, and reshape the intracellular or extracellular microenvironment of tumor-initiating cells to prepare for cell survival and amplification. However, there was significant heterogeneity in the changes of TRF2 in different types of tumor cells, and the level of TRF2 decreased significantly in some tumor cells [47,106,107]. The mechanism of its occurrence may be related to the functional status of intracellular acting proteins and degrading enzymes, which needs to be further studied. This also poses a challenge for the promotion of the strategy.
In conclusion, the presence of telomere TRF2 plays an important role in the survival and cell cycle maintenance of tumor-initiating cells. Since TRF2 knockout can terminate the process of somatic transformation into tumor-initiating cells and eliminate the process of somatic mutation, it seems possible to achieve the goal of completely preventing or eliminating tumor-initiating cell transformation by controlling the level of TRF2 in cells at different stages. Analysis, detection or alteration of the function and content of TRF2 in cells can provide promising research ideas for the early prevention and treatment of tumor-initiating cells [108,109].
The tumor-promoting stage (the second stage) and the environmental tumor-promoting factor effect of TRF2
The formation of tumor bodies often requires the transformation and construction of intracellular and extracellular microenvironments suitable for the growth of tumor initiation cells with the assistance of environmental tumor promoters, so that tumor initiation cells can colonize, grow and proliferate, and gradually form tumor bodies and produce pathological consequences [110-112]. These environmental tumor promoters usually do not cause revariation of the genetic material of the cell. The intracellular and extracellular microenvironment suitable for the survival of tumor-initiating cells is a plastic and multi-factor affected microecology, which is the product of long-term transformation. Although at this stage will not cause mass destructive to the tumor cell genes and chromosomes, but in epigenetics, transcriptome, tumor signaling pathways (including the activation of proto-oncogenes or inhibition of tumor suppressor genes), endocrine, immune regulation, and tumor angiogenesis and so on many aspects, internal and external microenvironment of cell is still in constant change [113-116]. This stage belongs to the tumor promotion stage, and its influencing factors are called tumor environmental promotion factors in this paper. For example, the classic cancer promoter TPA affects the transcription of specific genes in tumor-initiating cells by regulating cell protein kinase C, AP-1 and other cellular proteins, and induces the expression of c-fos and c-myc genes that are positively correlated with tumorgenesis. At the same time, the expression of Rb and α1-I3 genes negatively correlated with tumorigenesis was decreased. The ability to increase the frequency of gene amplification is more conducive to tumor formation and development [117-119].
Tumor formation is the process of proliferation, differentiation and infiltration of tumor-initiating cells. After the formation of initial cells with tumor karyotype, they are generally in a dormant state, long time latent in the body, they need appropriate environmental stimulation to promote their rapid development. The interaction between tumor-initiating cells and the microenvironment determines whether tumor-initiating cells continue to proliferate [120-122]. A number of studies have found that in a suitable microenvironment for tumor cell growth, the activation of various pro-tumor signaling pathways in tumor cells, the suppression of local immune function in tumor microenvironment and the rapid generation of tumor blood vessels are the three essential factors determining tumor formation and rapid development [123-125]. In addition to the telomere function related to somatic mutagenesis mentioned above, TRF2 protein in tumor-initiation cells plays an important role in the modification of the microenvironment conducive to tumor development among other functions besides telomere function of TRF2. The mechanism is closely related to the above three essential factors affecting tumor development in the microenvironment. Therefore, TRF2 also has obvious characteristics of environmental tumor-promoting factors.
Intracellular tumor-related signal transduction can induce up-regulation of TRF2 level and enhance the survival ability of tumor cells.
The transduction of tumor-related signals maintains and promotes the growth, development, proliferation, differentiation and environmental response of tumor cells, which is an indispensable element for the survival of tumor cells. Studies have found that Wnt/β-catenin, RAS/RAF/MEK/ERK, IKK/NF-ĸB and other tumor-related signal transduction pathway activation, leading to significantly higher intracellular TRF2 level [126-128].
The potential regulatory region downstream of the transcription start site of human TRF2 gene (TERF2) contains six TCF-LEF transcription factor binding sites (named Reg region). Tumor cell membrane receptors are stimulated by Wnt protein, and β-catenin nuclear translocation binds to TCF-LEF transcription factor, which specifically induces the activation of natural Reg region and increases the expression of TERF2. If the expression of β-Catenin Gene (Ctnnb1) is inhibited and β-catenin signal transduction is absent, TRF2 protein will decrease accordingly and Telomere Dysfunction Induced Foci (TIF) in Telomere region can be triggered. This was accompanied by a decrease in cell viability and an increase in the percentage of cells expressing β -galactosidase, a marker of cell senescence [129]. The Serine 323 (S323) residue of TRF2 is embedded with the consensus PXSP phosphorylation motif in MAPKs in different cell types, including normal and cancer cells. Erk1/2 and TRF2 interact in the cytoplasm, and the S323 residue of TRF2 can be phosphorylated by ERk1/2, increasing the half-life and stability of TRF2, participating in telomere protection, and promoting tumor growth by maintaining TRF2 levels [130].
Therefore, various tumor signal transduction pathways ultimately promote the expression of TRF2 gene or improve the stability of TRF2 protein, which is conducive to the accumulation of TRF2 in cells. Although the distribution level of TRF2 is different in different stages of tumor cells, and even decreased in the telomere region, TRF2 is an important protein molecule that tumor cells rely on for survival and cannot be replaced. Through cell signal transduction pathway, TRF2 establishes communication relationship with extracellular microenvironment. By sensing changes in external signals, TRF2 can enhance the environmental adaptability of tumor cells by adjusting epigenetic or protein interaction; On the other hand, it can regulate the expression of proteins and RNA to modify the peripheral microenvironment of tumors by autocrine or paracrine. Thus, TRF2 plays an important role as a non-telomere tumor-promoting factor [126-128]. The increase of the overall level of TRF2 in cells can also cooperate with cell cycle regulatory proteins to promote the mitosis of tumor cells, reduce the cell death caused by mitosis arrest, and produce the tolerance of tumor treatments, such as radiotherapy and chemotherapy. It has important synergistic effect on improving tumor cell viability [126,127,128]. Mechanically, the increase of TRF2 level can promote the binding of TRF2 to telomere or Interstitial Telomere DNA Sequence (ITS), protect related structures from various proteases or cytotoxic drugs, and make them more stable. The survival adaptability of tumor initiation cells was greatly improved. In this way, it is beneficial for tumor cells to colonize in the body tissue and play the role of external microenvironment modification. Overall, maintenance of TRF2 function is a prerequisite for significant transformation of the internal and external microenvironment of tumor cells and lays the foundation for accelerated cell expansion [131-133].
TRF2 promotes tumor angiogenesis and provides nutrition for rapid tumor expansion
Angiogenesis is an important manifestation of malignant tumor progression. Some studies have found that TRF2 is expressed in blood vessels in various tissues of human body [134- 136]. It is the transcriptional target of Wilms tumor suppressor WT1 and is critical for endothelial cell proliferation, migration, and tubular formation [137]. TRF2 gene knockdown can prevent vascular tissue formation and promote local fibrosis, inflammation and tumor necrosis. Sh RNA down regulation of TRF2 can disarrange the vascular network of tumor, and disperse endothelial cells and pericytes, showing the characteristics of fibrosis bands. Inflammation and extra vasation of red blood cells were also observed around tumor vessels after TRF2 knockout, indicating acute inflammation and vascular network disorder. Its mechanism of action is: TRF2 binds to and trans-activates the gene promoter of tumor endothelial angiogenic factor, Platelet Derived Growth Factor Receptor β (PDGFRβ), which increases PDGFRβ on tumor endothelial cell membrane, facilitating the formation of tumor vascular network and promoting tumor growth. This effect is independent of telomere function of TRF2 [137,138]. TRF2 also significantly affects VEGF-A levels in the secretory bodies of tumor cells, with a high positive correlation between TRF2 and VEGF-A promoting endothelial cell differentiation and angiogenesis [139,140]. This is due to the fact that TRF2 promotes the expression of sulf2 through binding to the distal regulatory element of the gene. Sulf2, a gene encoding endoglucosamine 6-sulfatase, is known to remove sulfates from the 6-O site of Heparan Sulfate (HS) [141] and affect tumor secretors [141]. By this mechanism, TRF2 attenuates the ability of Heparan Sulfate Proteoglycan (HSPG) to bind and isolate signaling molecules containing the heparin binding domain, including the angiogenic factor VEGF-A, making it more easily secreted into the Tumor Micro Environment (TME). This has a profound effect on tumor vascularization [140]. The promoting effect of TRF2 on tumor angiogenesis improves the poor living conditions in tumor microenvironment to a certain extent, and is more conducive to the crazy proliferation of tumor cells, resulting in the rapid enlargement of tumor body and serious clinical pathological response. In conclusion, clinical intervention of intracellular TRF2 level may be a promising tumor therapy.
Table 1: Effect of TRF2 silencing on expression levels of angiogenic and inflammatory genes in CAL33 cells, CAL27cells and CAL33 tumor xenografts.
| CAL33 | CAL27 | ||||||
|---|---|---|---|---|---|---|---|
| Genes | shC | sh1 | sh2 | shC | sh1 | sh2 | |
| CELLS | 36B4 | 100 | 100 | 100 | 100 | 100 | 100 |
| m-RPLP0 | 100 | 100 | 100 | 100 | 100 | 100 | |
| GADPH | 100 | 100 | 100 | 100 | 100 | 100 | |
| TERF2 | 100 | 70(*) | 40(***) | 100 | 62(*) | 32(***) | |
| CXCL1 | 100 | 326(***) | 307(***) | 100 | 129 | 80 | |
| CXCL7 | 100 | 110 | 131 | 100 | 199(*) | 379(***) | |
| CXCL8 | 100 | 357(***) | 197(***) | 100 | 212(*) | 142(*) | |
| CXCL9 | 100 | 235(**) | 334(***) | 100 | 293(*) | 355(**) | |
| CXCL10 | 100 | 162(*) | 479(***) | 100 | 217(**) | 267(**) | |
| IL6 | 100 | 400(***) | 697(***) | 100 | 289(***) | 100 | |
| PDGF-BB | 100 | 122(*) | 150(***) | 100 | 130 | 93 | |
| RANTES | 100 | 270(***) | 310(***) | 100 | 97 | 86 | |
| VEGF | 100 | 85 | 68(***) | 100 | 109 | 82 | |
| TUMORS | 36B4 | 100 | 100 | 100 | |||
| TERF2 | 100 | 91(*) | 51(***) | ||||
| CXCL1 | 100 | 276(***) | 302(**) | ||||
| CXCL7 | 100 | 124 | 125 | ||||
| CXCL8 | 100 | 96 | 119 | ||||
| CXCL10 | 100 | 179(*) | 1261(*) | ||||
| IL6 | 100 | 139 | 193 | ||||
| RANTES | 100 | 518(***) | 1325(**) | ||||
| VEGF | 100 | 48(*) | 57(**) | ||||
The percentage expression of the different genes evaluated by qPCR is shown. For the measured genes, the reference values [100] correspond to the content of a given gene in shC cells. The statistically significant differences are shown (ANOVAtest). *p< 0.05: **p< 0.01: ***p< 0.001

TRF2 inhibits inflammation and immune cell function in TME
In human malignant diseases, elevated TRF2 levels have a significant effect on tumor cell growth factor secretion [109]. In oral squamous cell carcinoma CAL33 and its metastases, knockdown TRF2 induced the expression of CXCL1, CXCL8, CXCL9, CXCL10, Interleukin-6 (IL6), PDGF-BB and RANTES, while decreased VEGF expression (109,142). As shown in Table 1, references were cited [109].
Cytokine expression Changes caused by TRF2 inhibition are thought to be associated with senescence related secretory phenotypes [143]. Beneficial to the elimination of senescent or diseased cells [143]. IL6, IL8/CXCL8, RANTES/CCL5 and GRO/CXCL1 are pro-inflammatory factors that cause local immune activation and inflammation [144-147]. The high expression of TRF2 in tumor cells can reduce the function of immune cells and regulatory factors in tumor TME, and help tumor cells evade immune surveillance. This is a very key step in the formation of human tumor body and plays an important role in the process of tumor expansion. In addition, molecular mechanism studies have shown that TRF2 can promote the expression of HSPG gene, which plays a very important role in the formation of exopolysaccharide calyx. TRF2, as a general remodeling agent of calyx structure, forms a microgradient structure that facilitates the recruitment of bone Marrow Derived Inhibitory Cells (MDSC) [148]. MDSC plays an important role in tumor angiogenesis, tumor cell survival, tumor metastasis, and premetastasis microenvironment formation [149]. The main immunosuppressive factors of MDSC include Indoleamine 2, 3-Dioxygenase (IDO), Arginase1 (ARG1), Reactive oxygen species (ROS), Interleukin-10, Tumor growth factor -β, Inducible Nitric Oxide Synthase (iNOS), COX-2, Nitric Oxide (NO), etc. [150]. At the same time, MDSC can also recruit regulatory T cells (Tregs) into the tumor microenvironment and inhibit the function of effector T cells (Teff) by secreting inhibitory factors such as Il-10, Il-35 and Transforming Growth Factor -β (TGF-β) [151-153]. The accumulation of MDSC in TME inhibits the recruitment and activation of Natural Killer Cells (NK), and also blocks the immune killing function of CD8+ T cells. In this microenvironment, the body loses the ability of immune surveillance and clearance, promoting the revival of dormant tumor-initiating cells and accelerating the proliferation of tumor cells [142,148,154]. Immune Checkpoint Blockers (ICBs) have been widely used in clinical cancer treatment, and have achieved remarkable clinical therapeutic effects, but at the same time, unwanted drug resistance has been generated. The mechanisms of drug resistance include checkpoint replacement mechanism, antigen presentation disorder, intracellular and extracellular metabolic disorder, abnormal tumor signaling pathway, immunosuppression of tumor microenvironment, transcriptome and epigenetic influence, etc. [155]. The immunosuppressive effect of TRF2 significantly affected the therapeutic effect of ICB [156]. Abnormal TRF2 may be an important factor of ICB drug resistance, and reasonable regulation of TRF2 level can provide a new idea for the selection of clinical treatment in the future.
Based on the above remodeling relationship between TRF2 and the survival microenvironment of tumor cells, we can consider the non-telomere function of TRF2 protein as an intracellular environmental tumor-promoting factor. Over expression of TERF2 gene or improvement of stability of TRF2 protein can rapidly activate changes in the microenvironment around tumor-initiating cells, recruit and transform tumor-related cells conducive to tumor cell growth, and have synergistic effects with various secreted cytokines and chemokines. Inhibit immune function, construct tumor angiogenesis, provide tumor cells with necessary nutrients, and finally break the dormancy of tumor initiation cells, and initiate tumor proliferation and development.
Please refer to Figure 1 for a schematic diagram of tumor formation and the tumor induction and environmental pro-tumor effects of TRF2
Discussion
TRF2 protein plays a key role in the process from somatic mutation to tumorigenesis. It is a common functional control protein in all stages of tumor formation and its function is closely related to tumor formation. High or low abnormal expression of TRF2 can lead to telomere protection loss or non-telomere dysfunction, which can lead to chromosome instability and induce fatal tumor diseases. In fact, TRF2 is not only over expressed in multiple tumor tissues [157-159], but also decreased in other tumor types [160-162], which is characterized by tumor heterogeneity. In terms of the mechanism of tumor cell formation, whether TRF2 levels are abnormally elevated or decreased in telomeres, severe telomere dysfunction can be induced. The over expression of TRF2 leads to the formation of overly compact DNA protein complexes, which reduces the binding of TRF1 protein, which promotes telomere replication, to telomere, thus impeding the advance of telomere replication fork, resulting in the stagnation of telomere DNA replication. Some telomeres are lost or shortened by end onuclease. The breakdown of telomere Ultrafine Anaphase Bridge (UFB) leads to random loss of large segments of telomere sequences and subtelomere regions, resulting in chromosome instability and mutations [162,163]. The reduction or loss of TRF2 destroys the T-loop structure and leaves the telomere ends unprotected. Due to lack of TRF2 regulation, endonuclease enzymes such as SLX4 and GEN1 and MUS81 can lead to catastrophic telomere excision and also to chromosome instability [164]. It can be seen that in terms of telomere functional integrity, too high or too low TRF2 in telomere can lead to chromosome instability and induce the generation of tumor-initiating cells at the stage of somatic mutation. This may provide an explanation for the formation of some heterogenous tumors with high or low TRF2 expression in clinic. In the stage of tumor formation, as mentioned above, TRF2, as an environmental tumor-promoting factor outside the telomere, has a tumor-promoting effect due to over expression, and is an important factor in promoting tumor development. In some tumors with reduced TRF2 levels, changes in the distribution and concentration of TRF2 in different parts of the nucleus may be responsible for maintaining and enhancing its tumor-promoting effect. In addition, there may also be synergistic effects of other endogenous and exogenous environmental pro-tumor factors, which need to be further studied. Due to the irreversibility of chromosomal aberrations and gene mutations, most of the current innovative tumor therapies focus on the regulation of signaling proteins in the tumor-promoting stage, such as tumor molecular targeting, tumor immune targeting, endocrine therapy, etc. If the function of TRF2 outside the telomere can be effectively regulated in tumor-initiating cells, the immunosuppression state and the formation of neovascularization in the tumor microenvironment can be blocked, so as to prevent local colonization of initiating tumor cells and achieve the blocking effect of tumor body formation. Combined with the existing clinical tumor treatment methods, the transformation of tumor initiation cells was eliminated, and the overall survival rate and prognosis of tumor patients were improved. Therefore, how to rationally apply new agents to control the function of TRF2 in tumor cells has potential significance for the development of new ideas for clinical treatment of tumor. However, the tumor-specific targeting of TRF2 will still face greater challenges. How to get rid of the influence of TRF2 on normal cells and reduce adverse reactions remains to be further studied.
Conclusion
At present, the theories of tumor formation have their merits. Among them, the aneuploidy theory and gene mutation theory explain the transformation process of somatic tumor. In recent years, many studies have found that these two theories do not exist in isolation. Gene mutations and chromosomal aberrations in somatic cells complement each other and both are indispensable processes leading to the formation of tumor transformed cells. Both can be the primary cause of tumour formation, in which the transformation of somatic cells into tumours is caused by chromosomal aberrations or gene mutations. When tumor-initiating cells eventually form, there must be aneuploidy and a large number of genetic mutations. TRF2 protein is involved in all of these processes, so we believe that TRF2 is a very important factor in tumor formation. The functional status of this protein can affect both chromosome and gene stability. Abnormal telomere or non-telomere function of TRF2 can lead to chromosome instability, abnormal gene expression, continuous chromosome aberration and gene mutation, and promote the formation of mutated tumor transformed cells. In addition, the extracellular function of TRF2 can promote the formation of tumor microenvironment, which is conducive to the colonization and proliferation of tumor-initiating cells and tumor body formation. Targeting TRF2 provides a new idea for tumor prevention and treatment, and has far-reaching application value and significance.
Declarations
Ethics approval and consent to participate: Not applicable.
Consent for publication: All authors made substantial contributions to the manuscript and gave their approval on the final version. The present publication has not been published in this form previously and is not under consideration for publication elsewhere and agree to publish in this journal.
Availability of data and materials: All information or data in this review are from publicly published international journals.
Competing interests: The authors declare that they have no competing interests.
Funding: Sichuan Science and Technology Planning Project (Transformation of Scientific and Technological achievements in Scientific research institutions)NO.2022JDZH0014.
Authors’ contributions: These authors contributed equally.
Acknowledgements: Not applicable.
References
- Faguet GB. A brief history of cancer: Age-old milestones underlying our current knowledge database. Int J Cancer. 2015; 136: 2022-2036.
- Bouck N, di Mayorca G. Somatic mutation as the basis for malignant transformation of BHK cells by chemical carcinogens. Nature. 1976; 264: 722-727.
- Krontiris TG, Cooper GM. Transforming activity of human tumor DNAs. Proc Natl Acad Sci U S A. 1981; 78: 1181-1184.
- Cooper GM, Okenquist S, Silverman L. Transforming activity of DNA of chemically transformed and normal cells. Nature. 1980; 284: 418-421.
- Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015; 348: 880-886.
- Yokoyama A, Kakiuchi N, Yoshizato T, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019; 565: 312-317.
- Lee-Six H, Olafsson S, Ellis P, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019; 574: 532-537.
- Brunner SF, Roberts ND, Wylie LA, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019; 574: 538-542.
- Moore L, Leongamornlert D, Coorens THH, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020; 580: 640-646.
- Yoshida K, Gowers KHC, Lee-Six H, et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020; 578: 266-272.
- Kim SK, Takeda H, Takai A, et al. Comprehensive analysis of genetic aberrations linked to tumorigenesis in regenerative nodules of liver cirrhosis. J Gastroenterol. 2019; 54: 628-640.
- Zhu M, Lu T, Jia Y, et al. Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell. 2019; 177: 608-621.e12.
- Martincorena I, Fowler JC, Wabik A, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018; 362: 911-917.
- Goerttler K, Loehrke H, Schweizer J, Hesse B. Two-stage skin carcinogenesis by systemic initiation of pregnant mice with 7,12-dimethylbenz(a) anthracene during gestation days 6-20 and postnatal promotion of the F 1-generation with the phorbol ester 12-tetradecanoylphorbol-13-acetate. J Cancer Res Clin Oncol. 1980; 98: 267-275.
- Goerttler K, Loehrke H, Hesse B, Milz A, Schweizer J. Diaplacental initiation of NMRI mice with 7,12-dimethylbenzaanthracene during gestation days 6--20 and postnatal treatment of the F1-generation with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate: tumor incidence in organs other than the skin. Carcinogenesis. 1981; 2: 1087-1094.
- Mucci LA, Hjelmborg JB, Harris JR, et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries published correction appears in JAMA. 2016; 315: 822. JAMA. 2016; 315: 68-76.
- Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343: 78-85.
- Marx J. Debate surges over the origins of genomic defects in cancer. Science. 2002; 297: 544-546.
- Strunnikov VA, Uryvaeva IV, Brodskiĭ VIa. Dvumutatsionnaia gipoteza kantserogeneza Double-mutation hypothesis of carcinogenesis. Tsitol Genet. 1984; 18: 380-391.
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999; 400: 464-468.
- Pringle CR. The oncogene hypothesis and the search for human cancer virusus. Scott Med J. 1975; 20: 61-67.
- Caesar G. Analysis of the oncogene hypothesis. Med Hypotheses. 1984; 14: 379-385.
- Stenbäck F, Peto R, Shubik P. Initiation and promotion at different ages and doses in 2200 mice. I. Methods, and the apparent persistence of initiated cells. Br J Cancer. 1981; 44: 1-14.
- Loehrke H, Schweizer J, Dederer E, Hesse B, Rosenkranz G, Goerttler K, et al. On the persistence of tumor initiation in two-stage carcinogenesis on mouse skin. Carcinogenesis. 1983; 4: 771-775.
- Pfitzenmaier J, Ellis WJ, Hawley S, et al. The detection and isolation of viable prostate-specific antigen positive epithelial cells by enrichment: A comparison to standard prostate-specific antigen reverse transcriptase polymerase chain reaction and its clinical relevance in prostate cancer. Urol Oncol. 2007; 25: 214-220.
- Vessella RL, Pantel K, Mohla S. Tumor cell dormancy: an NCI workshop report. Cancer Biol Ther. 2007; 6: 1496-1504.
- Olaharski AJ, Sotelo R, Solorza-Luna G, et al. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis. 2006; 27: 337-343.
- Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007; 17: 157-162.
- Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y, et al. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol. 2007; 17: 431-437.
- Fodde R, Kuipers J, Rosenberg C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001; 3: 433-438.
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, et al. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001; 3: 429-432.
- Hirpara A, Bloomfield M, Duesberg P. Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes (Basel). 2018; 9: 402.
- Duesberg P, Li R, Fabarius A, Hehlmann R. The chromosomal basis of cancer. Cell Oncol. 2005; 27: 293-318.
- Endo Y, Watanabe M, Miyajima-Magara N, et al. DNA aneuploidy and centrosome amplification in canine tumor cell lines. Tissue Cell. 2019; 61: 67-71.
- Yao DW, Balanis NG, Eskin E, Graeber TG. A linear mixed model approach to gene expression-tumor aneuploidy association studies. Sci Rep. 2019; 9: 11944.
- Liu J, Wang L, Wang Z, Liu JP. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells. 2019; 8: 54.
- Rai R, Chen Y, Lei M, Chang S. TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat Commun. 2016; 7: 10881.
- Mendez-Bermudez A, Lototska L, Bauwens S, et al. Genome-wide Control of Heterochromatin Replication by the Telomere Capping Protein TRF2. Mol Cell. 2018; 70: 449-461.e5.
- Timashev LA, Babcock H, Zhuang X, de Lange T. The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes Dev. 2017; 31: 578-589.
- von Morgen P, Maciejowski J. The ins and outs of telomere crisis in cancer. Genome Med. 2018; 10: 89.
- Ishikawa F. Telomere crisis, the driving force in cancer cell evolution. Biochem Biophys Res Commun. 1997; 230: 1-6.
- Lin TT, Letsolo BT, Jones RE, et al. Telomere dysfunction and fusion during the progression of chronic lymphocytic leukemia: Evidence for a telomere crisis. Blood. 2010; 116: 1899-1907.
- Hayashi MT. Seikagaku. 2016; 88: 756-760.
- Bailey SM, Murnane JP. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006; 34: 2408-2417.
- Castro-Vega LJ, Jouravleva K, Liu WY, et al. Telomere crisis in kidney epithelial cells promotes the acquisition of a microRNA signature retrieved in aggressive renal cell carcinomas. Carcinogenesis. 2013; 34: 1173-1180.
- Giraud-Panis MJ, Pisano S, Poulet A, Le Du MH, Gilson E, et al. Structural identity of telomeric complexes. FEBS Lett. 2010; 584: 3785-3799.
- Salhab M, Jiang WG, Newbold RF, Mokbel K. The expression of gene transcripts of telomere-associated genes in human breast cancer: correlation with clinico-pathological parameters and clinical outcome. Breast Cancer Res Treat. 2008; 109: 35-46.
- Yamada K, Yagihashi A, Yamada M, et al. Decreased gene expression for telomeric-repeat binding factors and TIN2 in malignant hematopoietic cells. Anticancer Res. 2002; 22: 1315-1320.
- Blanco R, Muñoz P, Flores JM, Klatt P, Blasco MA, et al. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev. 2007; 21: 206-220.
- Matsutani N, Yokozaki H, Tahara E, et al. Expression of telomeric repeat binding factor 1 and 2 and TRF1-interacting nuclear protein 2 in human gastric carcinomas. Int J Oncol. 2001; 19: 507-512.
- Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, et al. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 2002; 277: 41110-41119.
- Stavropoulos DJ, Bradshaw PS, Li X, et al. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet. 2002; 11: 3135-3144.
- Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012; 148: 349-361.
- Sänger N, Effenberger KE, Riethdorf S, et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int J Cancer. 2011; 129: 2522-2526.
- Kim MY, Oskarsson T, Acharyya S, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009; 139: 1315-1326.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002; 3: 991-998.
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002; 3: 991-998.
- Hoover, R. N. in Origins of Human Cancer (eds. Hiatt, H. H.,Watson, J. D. & Winsten, J.A.) 369–379 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1977).
- Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994; 1: 447-456.
- Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998; 95: 7556-7561.
- Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood. 2001; 97: 192-197.
- Street SE, Trapani JA, MacGregor D, Smyth MJ. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J Exp Med. 2002; 196: 129-134.
- Rosenthal R, Cadieux EL, Salgado R, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019; 567: 479-485.
- Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019; 51: 202-206.
- Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019; 51: 202-206.
- Donehower LA, Soussi T, Korkut A, et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas published correction appears in Cell Rep. 2019; 28: 3010. Cell Rep. 2019; 28: 1370-1384.e5.
- Kuenzi BM, Ideker T. A census of pathway maps in cancer systems biology published correction appears in Nat Rev Cancer. 2021; 21: 212. Nat Rev Cancer. 2020; 20: 233-246.
- Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982; 297: 474-478.
- Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. 1982; 298: 343-347.
- Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986; 323: 643-646.
- Merlo A, Herman JG, Mao L, et al. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995; 1: 686-692.
- Powers MP. The ever-changing world of gene fusions in cancer: a secondary gene fusion and progression. Oncogene. 2019; 38: 7197-7199.
- Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458: 719-724.
- Balmain A. The critical roles of somatic mutations and environmental tumor-promoting agents in cancer risk. Nat Genet. 2020; 52: 1139-1143.
- Radomska KJ, Coulpier F, Gresset A, et al. Cellular Origin, Tumor Progression, and Pathogenic Mechanisms of Cutaneous Neurofibromas Revealed by Mice with Nf1 Knockout in Boundary Cap Cells. Cancer Discov. 2019; 9: 130-147.
- Drost J, van Jaarsveld RH, Ponsioen B, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015; 521: 43-47.
- Parsons BL. Many different tumor types have polyclonal tumor origin: Evidence and implications. Mutat Res. 2008; 659: 232-247.
- Lynch HT, Casey MJ, Lynch J, White TE, Godwin AK. Genetics and ovarian carcinoma. Semin Oncol. 1998; 25: 265-280.
- Xu Y. Induction of genetic instability by gain-of-function p53 cancer mutants. Oncogene. 2008; 27: 3501-3507.
- Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem. 2012; 113: 433-439.
- Woo RA, Poon RY. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 2004; 18: 1317-1330.
- Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997; 17: 231-235.
- Bilaud T, Brun C, Ancelin K, Koering CE, Laroche T, et al. Telomeric localization of TRF2, a novel human telobox protein. Nat Genet. 1997; 17: 236-239.
- Benarroch-Popivker D, Pisano S, Mendez-Bermudez A, et al. TRF2-Mediated Control of Telomere DNA Topology as a Mechanism for Chromosome-End Protection. Mol Cell. 2016; 61: 274-286.
- Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999; 97: 503-514.
- Greider CW. Telomeres do D-loop-T-loop. Cell. 1999; 97: 419-422.
- Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR 3rd, Denchi EL, et al. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature. 2013; 494: 502-505.
- Ribes-Zamora A, Indiviglio SM, Mihalek I, Williams CL, Bertuch AA, et al. TRF2 interaction with Ku heterotetramerization interface gives insight into c-NHEJ prevention at human telomeres. Cell Rep. 2013; 5: 194-206.
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012; 336: 593-597.
- Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002; 295: 2446-2449.
- Smogorzewska A, van Steensel B, Bianchi A, et al. Control of human telomere length by TRF1 and TRF2. Mol Cell Biol. 2000; 20: 1659-1668.
- Wilson JS, Tejera AM, Castor D, Toth R, Blasco MA, Rouse J, et al. Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep. 2013; 4: 853-860.
- Muñoz P, Blanco R, Blasco MA. Role of the TRF2 telomeric protein in cancer and ageing. Cell Cycle. 2006; 5: 718-721.
- Menendez JA, Rubio MA, Campisi J, Lupu R. Heregulin, a new regulator of telomere length in human cells. Oncotarget. 2015; 6: 39422-39436.
- Menendez JA, Benboudjema L, Vellon L, et al. Heregulin, a new interactor of the telosome/shelterin complex in human telomeres. Oncotarget. 2015; 6: 39408-39421.
- Kim W, Ludlow AT, Min J, et al. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016; 14: e2000016.
- Bloomfield M, Duesberg P. Is cancer progression caused by gradual or simultaneous acquisitions of new chromosomes?. Mol Cytogenet. 2018; 11: 4.
- Duesberg PH. Does aneuploidy destabilize karyotypes automatically?. Proc Natl Acad Sci U S A. 2014; 111: E974.
- Ermler S, Krunic D, Knoch TA, et al. Cell cycle-dependent 3D distribution of telomeres and Telomere Repeat-Binding Factor 2 (TRF2) in HaCaT and HaCaT-myc cells. Eur J Cell Biol. 2004; 83:681-690.
- Nijjar T, Bassett E, Garbe J, et al. Accumulation and altered localization of telomere-associated protein TRF2 in immortally transformed and tumor-derived human breast cells. Oncogene. 2005; 24: 3369-3376.
- Lajoie V, Lemieux B, Sawan B, et al. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood. 2015; 125: 2101-2110.
- Hussain T, Saha D, Purohit G, et al. Transcription regulation of CDKN1A (p21/CIP1/WAF1) by TRF2 is epigenetically controlled through the REST repressor complex. Sci Rep. 2017; 7: 11541.
- Hayashi MT, Cesare AJ, Rivera T, Karlseder J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature. 2015; 522: 492-496.
- Khadka P, Lee JH, Baek SH, Oh SY, Chung IK. DNA-PKcs-interacting protein KIP binding to TRF2 is required for the maintenance of functional telomeres. Biochem J. 2014; 46: 19-30.
- He X, Liao J, Liu F, et al. Functional repair of p53 mutation in colorectal cancer cells using trans-splicing. Oncotarget. 2015; 6: 2034-2045.
- Pascua I, Fernández-Marcelo T, Sánchez-Pernaute A, et al. Prognostic value of telomere function in gastric cancers with and without microsatellite instability. Eur J Gastroenterol Hepatol. 2015; 27: 162-169.
- Yamada K, Yagihashi A, Yamada M, et al. Decreased gene expression for telomeric-repeat binding factors and TIN2 in malignant hematopoietic cells. Anticancer Res. 2002; 22: 1315-1320.
- El Maï M, Janho Dit Hreich S, Gaggioli C, et al. A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers (Basel). 2021; 13: 2998.
- Benhamou Y, Picco V, Raybaud H, et al. Telomeric repeat-binding factor 2: A marker for survival and anti-EGFR efficacy in oral carcinoma. Oncotarget. 2016; 7: 44236-44251.
- Bradford JW, Baldwin AS. IKK/nuclear factor-kappaB and oncogenesis: Roles in tumor-initiating cells and in the tumor microenvironment. Adv Cancer Res. 2014; 121: 125-145.
- Tanno T, Lim Y, Wang Q, et al. Growth differentiating factor 15 enhances the tumor-initiating and self-renewal potential of multiple myeloma cells. Blood. 2014; 123: 725-733.
- Schwab LP, Peacock DL, Majumdar D, et al. Hypoxia-inducible factor 1α promotes primary tumor growth and tumor-initiating cell activity in breast cancer. Breast Cancer Res. 2012; 14: R6.
- Luo M, Yang F, Huang SX, et al. Two-stage model of chemically induced hepatocellular carcinoma in mouse. Oncol Res. 2013; 20: 517-528.
- Taniai E, Kawai M, Dewa Y, et al. Crosstalk between PTEN/Akt2 and TGF beta signaling involving EGF receptor down-regulation during the tumor promotion process from the early stage in a rat two-stage hepatocarcinogenesis model. Cancer Sci. 2009; 100: 813-820.
- Takahashi M, Shibutani M, Woo GH, et al. Cellular distributions of molecules with altered expression specific to the tumor promotion process from the early stage in a rat two-stage hepatocarcinogenesis model. Carcinogenesis. 2008; 29: 2218-2226.
- Oladipupo SS, Kabir AU, Smith C, Choi K, Ornitz DM, et al. Impaired tumor growth and angiogenesis in mice heterozygous for Vegfr2 (Flk1). Sci Rep. 2018; 8: 14724.
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982; 257: 7847-7851.
- Angel P, Imagawa M, Chiu R, et al. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987; 49: 729-739.
- Zhang Chunyan, Zhao Qingzheng, Wang Ping, et al. Effects of carcinogens on gene expression J. Journal of the Chinese Academy of Medical Sciences ,1995: 11-15
- White DE, Kurpios NA, Zuo D, et al. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004; 6: 159-170.
- Tigyi GJ, Yue J, Norman DD, et al. Regulation of tumor cell - Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv Biol Regul. 2019; 71: 183-193.
- Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015; 368: 7-13.
- Najafi M, Ahmadi A, Mortezaee K. Extracellular-signal-regulated kinase/mitogen-activated protein kinase signaling as a target for cancer therapy: an updated review. Cell Biol Int. 2019; 43: 1206-1222.
- Giraldo NA, Sanchez-Salas R, Peske JD, et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019; 120: 45-53.
- Fabian KL, Storkus WJ. Immunotherapeutic Targeting of Tumor-Associated Blood Vessels. Adv Exp Med Biol. 2017; 1036: 191-211.
- Diala I, Wagner N, Magdinier F, et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep. 2013; 14: 356-363.
- Picco V, Coste I, Giraud-Panis MJ, Renno T, Gilson E, Pagès G, et al. ERK1/2/MAPK pathway-dependent regulation of the telomeric factor TRF2. Oncotarget. 2016; 7: 46615-46627.
- An J, Wu M, Xin X, et al. Inflammatory related gene IKKα, IKKβ, IKKγ cooperates to determine liver cancer stem cells progression by altering telomere via heterochromatin protein 1-HOTAIR axis. Oncotarget. 2016; 7: 50131-50149.
- Diala I, Wagner N, Magdinier F, et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep. 2013; 14: 356-363.
- An J, Wu M, Xin X, et al. Inflammatory related gene IKKα, IKKβ, IKKγ cooperates to determine liver cancer stem cells progression by altering telomere via heterochromatin protein 1-HOTAIR axis. Oncotarget. 2016; 7: 50131-50149.
- Pedroso IM, Hayward W, Fletcher TM. The effect of the TRF2 N-terminal and TRFH regions on telomeric G-quadruplex structures. Nucleic Acids Res. 2009; 37: 1541-1554.
- Smith ED, Garza-Gongora AG, MacQuarrie KL, Kosak ST. Interstitial telomeric loops and implications of the interaction between TRF2 and lamin A/C. Differentiation. 2018; 102: 19-26.
- Mignon-Ravix C, Depetris D, Delobel B, Croquette MF, Mattei MG, et al. A human interstitial telomere associates in vivo with specific TRF2 and TIN2 proteins. Eur J Hum Genet. 2002; 10: 107-112.
- Wang J, Uryga AK, Reinhold J, et al. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation. 2015; 132: 1909-1919.
- Morgan RG, Walker AE, Trott DW, et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J Mol Cell Cardiol. 2019; 127: 74-82.
- Spyridopoulos I, Haendeler J, Urbich C, et al. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation. 2004; 110: 3136-3142.
- El Maï M, Wagner KD, Michiels JF, et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014; 9: 1047-1060.
- Maï ME, Wagner KD, Michiels JF, Gilson E, Wagner N. TRF2 acts as a transcriptional regulator in tumor angiogenesis. Mol Cell Oncol. 2015; 2: e988508.
- Dinami R, Porru M, Amoreo CA, et al. TRF2 and VEGF-A: an unknown relationship with prognostic impact on survival of colorectal cancer patients. J Exp Clin Cancer Res. 2020; 39: 111.
- Zizza P, Dinami R, Porru M, et al. TRF2 positively regulates SULF2 expression increasing VEGF-A release and activity in tumor microenvironment. Nucleic Acids Res. 2019; 47: 3365-3382.
- Ai X, Do AT, Kusche-Gullberg M, Lindahl U, Lu K, Emerson CP Jr. Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem. 2006; 281: 4969-4976.
- Biroccio A, Cherfils-Vicini J, Augereau A, et al. TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat Cell Biol. 2013; 15: 818-828.
- Pérez-Mancera PA, Young AR, Narita M. Inside and out: The activities of senescence in cancer. Nat Rev Cancer. 2014; 14: 547-558.
- Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014; 2014: 292376.
- Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015; 36: 92-101.
- Zarogoulidis P, Katsikogianni F, Tsiouda T, Sakkas A, Katsikogiannis N, Zarogoulidis K, et al. Interleukin-8 and interleukin-17 for cancer. Cancer Invest. 2014; 32: 197-205.
- Verbeke H, Struyf S, Laureys G, Van Damme J. The expression and role of CXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. 2011; 22: 345-358.
- Cherfils-Vicini J, Iltis C, Cervera L, et al. Cancer cells induce immune escape via glycocalyx changes controlled by the telomeric protein TRF2. EMBO J. 2019; 38: e100012.
- Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013; 13: 739-752.
- Groth C, Hu X, Weber R, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. 2019; 120: 16-25.
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008; 133: 775-787.
- Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC, et al. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003; 170: 1240-1248.
- Oida T, Zhang X, Goto M, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. 2003; 170: 2516-2522.
- Simonet T, Zaragosi LE, Philippe C, et al. The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res. 2011; 21: 1028-1038.
- Wang Z, Wu X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020; 9: 8086-8121.
- Ilié M, Lantéri E, Chamorey E, et al. Association of TRF2 expression and myeloid-derived suppressor cells infiltration with clinical outcome of patients with cutaneous melanoma. Oncoimmunology. 2021; 10: 1901446.
- Blanco R, Muñoz P, Flores JM, Klatt P, Blasco MA, et al. Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes Dev. 2007; 21: 206-220.
- Matsutani N, Yokozaki H, Tahara E, et al. Expression of telomeric repeat binding factor 1 and 2 and TRF1-interacting nuclear protein 2 in human gastric carcinomas. Int J Oncol. 2001; 19: 507-512
- Oh BK, Kim YJ, Park C, Park YN. Up-regulation of telomere-binding proteins, TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am J Pathol. 2005; 166: 73-80.
- Salhab M, Jiang WG, Newbold RF, Mokbel K. The expression of gene transcripts of telomere-associated genes in human breast cancer: correlation with clinico-pathological parameters and clinical outcome. Breast Cancer Res Treat. 2008; 109: 35-46.
- Yamada K, Yagihashi A, Yamada M, et al. Decreased gene expression for telomeric-repeat binding factors and TIN2 in malignant hematopoietic cells. Anticancer Res. 2002; 22: 1315-1320
- Nera B, Huang HS, Lai T, Xu L. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun. 2015; 6: 10132.
- Rai R, Zheng H, He H, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010; 29: 2598-2610.
- Saint-Léger A, Koelblen M, Civitelli L, et al. The basic N-terminal domain of TRF2 limits recombination endonuclease action at human telomeres. Cell Cycle. 2014; 13: 2469-2474.