
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Pancytopenia, a rare case report of epimerase deficiency galactosemia
Reihaneh Mohsenipour1; Nejat Mahdieh1,3; Parastoo Rostami4*
1Growth and Development Research Center, Children’s Medical Center, Tehran University of Medical Sciences, Tehran, Iran.
2Cardiogenetic Research Centre, Rajaie Cardiovascular Medical and Research Centre, Iran University of Medical Sciences, Tehran, Iran.
3Growth and Development Research Center, Department of Endocrinology and Metabolism, Pediatric Center of Excellence, Tehran University of Medical Sciences, Tehran, Iran.
*Corresponding Author : Parastoo Rostami
Department of Endocrinology and Metabolism Children’s Medical Center, Keshavarz Blvd, Tehran, Iran.
Email: Drp_rostami@yahoo.com
Received : Aug 16, 2023
Accepted : Sep 06, 2023
Published : Sep 13, 2023
Archived : www.jcimcr.org
Copyright : © Rostami P (2023).
Abstract
Galactose is the most important sugar in dairy products. The liver and kidney are the main organs involved in galactose metabolism. There are three enzymes involved in the galactose metabolism pathway, the deficiency of UDP-galactose-4-epimerase is associated with galactosemia type III. A benign form of GALE deficiency is only found in the erythrocyte and leucocytes but the presentations of the generalized form are the same as classic galactosemia. Herein we’ve reported two siblings with a novel pathogenic variant, c.1002G > T at the protein level as p.Trp334Cys in the GALE gene who presented with pancytopenia.
Keywords: Pancytopenia; Galactosemia; Newborn screening; Case report.
Citation: Mohsenipour R, Mahdieh N, Rostami P. Pancytopenia, a rare case report of epimerase deficiency galactosemia. J Clin Images Med Case Rep. 2023; 4(9): 2588.
Introduction
Metabolism of galactose
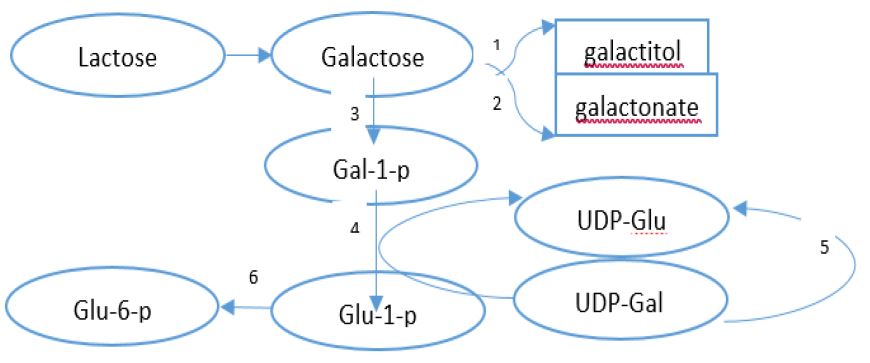
Carbohydrates are the most common precursors of production of the energy in the body. Among monosaccharides, only glucose is stored as glycogen in the liver and muscles [1]. After glucose, galactose and fructose are used as an alternative sources of energy in the body. There is a small amount of galactose in the foods, whilst there is a bunch of galactose in the milk and dairy products in the form of lactose [2,3]. Lactose is hydrolyzed into glucose and galactose by lactase in the intestinal, afterwards, galactose is absorbed through a Sodium/Glucose-galactose cotransporter (SGLT1) [1]. All enzymes which are involved in galactose metabolism are located in the liver. The pathway of galactose metabolism (Leloir pathway) in the liver is shown in figure 1.
Classification of galactosemia
Classic galactosemia or Type 1 (#230400), is due to Galactose-1 phosphate uridyltransferase (GALT) deficiency which is the most common and most severe of galactosemia. Deficiency of Galactose Kinase (GALK) leads to type II galactosemia (#230200) and finally, galactosemia type III (#230350) is caused by UDP-galactose-4-epimerase deficiency is more uncommon than the other variants. All disorders are inherited as an autosomal recessive pattern. GALE enzyme is responsible for two steps in the Leloir pathway of galactose metabolism: conversion of UDP-galactose (UDP-gal) to UDP-glucose (UDP-glc) and formation of UDP-N-acetylgalactosamine (UDP-galNAc) from UDP-N-acetylglucosamine (UDP-glcNAc) [1,4].
Prevalence and clinical manifestations of epimerase deficiency galactosemia
The exact prevalence the galactosemia type III has unknown yet but epimerase deficiency galactosemia found by newborn screening is nearly 1:6,700 in African American infants and about 1:70,000 the US infants of European ancestry [5,6-21]. Clinical manifestations of GALE deficiency are divided into three categories: The generalized form of GALE deficiency is very rare and symptoms are very severe resembling classic galactosemia. Patients with an intermediate form of epimerase deficiency galactosemia are at risk for long-term complications such as vomiting, hypoglycemia, seizure, learning disability, and cataracts. Peripheral or benign epimerase deficiency galactosemia is restricted to red and white blood cells and the majority of patients are asymptomatic.
Laboratory findings of epimerase deficiency galactosemia
Similar to classic galactosemia the level of galactose, galactitol, Gal-1P, and UDPgal in blood are elevated in the epimerase deficiency but GULT activity is normal. The high level of total galactose (galactose and galactose-1-phosphate) is detectable through newborn screening. Molecular analysis or enzyme activity in the first trimester of pregnancy by chorionic villus biopsy is recommended in families with a similar disease in other siblings.
Treatment of galactosemia
The mainstay of treatment for all types of galactosemia is the galactose restriction diet but isolated peripheral GALE deficiency does not require galactose restriction. Galactose should be eliminated from the diet as soon as galactosemia is suspected. In infants a soy-based formula such as ProSobee, Nursoy, Alsoy and Isomil are appropriate. Similar to patients with classic galactosemia, patients with generalized epimerase deficiency respond well to dietary restriction of galactose, which prevents the potentially lethal acute symptoms, although long-term complications may persist [2,22-29].
Clinical report
A 12-year-old male who referred to a Gastroenterology clinic because of scleral icterus at the age of 9 years old. He is the first child from consanguine parents. By the age of nine, he was perfectly healthy but his parents noticed icterus in his child’s eyes. At that time physical examinations were normal expect scleral icterus and on the laboratory tests, he had hyperbilirubinemia [total bilirubin: 3.4 mg/dl (normal: 0.3-1.2 mg/dl), direct bilirubin: 0.51 mg/dl (normal: 0-0.5 mg/dl)], but liver transaminases were normal. Gilbert syndrome has been diagnosed by a gastroenterologist. During periodic monitoring for icterus, pancytopenia [WBC count: 3.39 × 103/ μL (normal: 6-10 × 103/ μL), hemoglobin: 9.4 gr/dl (normal: 11.5-15.5 gr/dl), platelet: 90 × 103/ μL (normal: 170-450 × 103/ μL)] was revealed. Other tests including; G6PD, Alkp, LDH, HBS Ag, HCV Ab, HIV PCR, direct and indirect coombs, serum amino acids profile, and alpha-fetoprotein, were normal. Pelvic and abdomen Solography imaging and bone marrow aspiration/biopsy were normal. Whole exome sequencing for more evaluation of pancytopenia and icterus was performed and GALE deficiency appeared. According to this diagnosis, serum galactose, and galactose-1-phosphate level was assessed, and all completely were normal. His patient’s sister, mother, and father were assessed. Her sister had just thrombocytopenia at the age of 3 years old; platelet: 126000, but RBC and WBC count and Hb were normal. After 6 months she experienced leukopenia (WBC = 3800 × 103/ μL), thrombocytopenia (platelet = 108000 × 103/ μL) and anemia (Hb = 9.6 gr/dl). As his brother, the liver transaminases, and serum galactose galactose-1-phosphate were normal. Despite normal serum galactose and galactose-1-phosphate level, an experimental galactose restriction diet was started for them but WBC, RBC and platelet count, serum Hb level did not change after one year, therefore the galactose-free diet was discontinued. During the follow-up of patients, after two months of discontinuation of the galactose-free diet, pancytopenia became more severe and we had to continue their diet. The boy developed splenomegaly after two years of monitoring, which seems to be caused by compensatory extra medullary hematopoiesis.
Genetic investigations
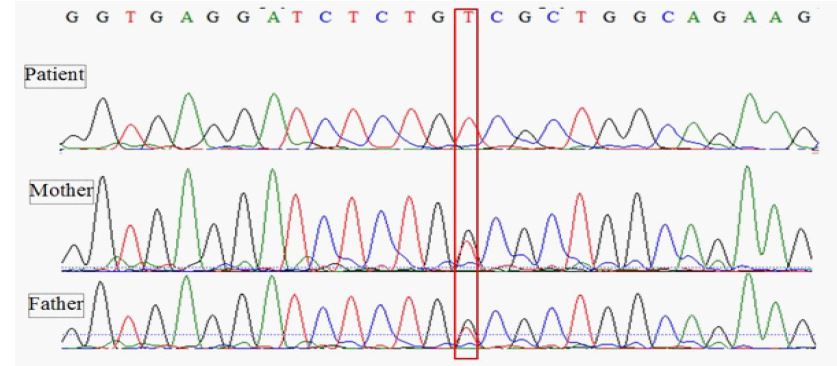
Genetic counseling and pedigree drawing were done in the affected families. DNA was extracted according to standard protocols. Whole exome sequencing (WES) was performed to find the causal variants. The coding region’s enrichment was performed by the Twist Human Core Exome Kit. Sequencing was done on the Illumina (NovaSeq™6000 Sequencing System platform) with an average coverage depth of about 100x. Bioinformatics analysis was applied according to standard pipelines described elsewhere [10]. Briefly, quality control of the reads was done using the FastQC tool (version 0.11.9) [11]. The reads were aligned to the reference genome of humans (GRCh38/hg38) using the Bowtie2 (Version 2.4.0) aligning tool [12]. Picard was used to convert SAM files to BAM (Binary Alignment Map) files [13]. The genome analysis toolkit (GATK) was used for local realignment of insertion/deletion (indels) [14]. wANNOVAR (http://wannovar.wglab.org/) was applied for annotating the variants [15]. Variants with a minor allele frequency (MAF) of more than 1% were removed. The following databases were used to compare the variants: Exome Sequencing Project 6500 (http://evs.gs.washington.edu/EVS/), the Exome Aggregation Database (http://exomad.broadinstitute.org/), the Exome Aggregation Consortium database (http://exac.broadinstitute.org) and the Greater Middle East Variome Project (http://igm.ucsd.edu/gme/). Other available software tools applied to predicting the pathogenicity of the variant were as follows Sorting Intolerant From Tolerant (https://sift.bii.a-star.edu.sg) and PolyPhen-2 (http://genetics.bwh. harvard.edu/pph2/index.shtml). Moreover, this variant had a Combined Annotation Dependent Depletion (CADD) score of 23 (https://cadd.gs.washington.edu) that is highly pathogenic. WES of the patient and his younger sister showed a homozygous mutation in GALE (NM_001127621; exon 1: c.1002G > T) coding for UDP-Galactose-4-Epimerase. This mutation causes a missense amino acid change at highly conserved codon 334 (p.W334C). This mutation has not been reported in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/all.php) and ClinVar Miner (https://clinvarminer.genetics.utah.edu). The parents were heterozygous for the same (Figure 2).
Discussion
The first case of GALE deficiency was reported through new born screening [1]. UDP-galactose-4-epimerase encodes by GALE enzyme, which is essential to galactose metabolism/production and glycosylation of proteins and lipids [2,3]. Since 1972 till now, about 150 patients of galactosemia type III with different clinical manifestations based on the severity of epimerase deficiency and through newborn screening (about 92 patients) have been reported in the worldwide [2,4,6-39]. Benign variant of GALE deficiency restricted to peripheral red and white blood cells but the generalized variant is very rare resulting from GALE deficiency in many tissues [1,2]. Inadequate GALE activity leads to defect of glycosylation that effect on hematopoiesis processes. Glycosylation of the proteins and lipids for hematopoiesis and platelet synthesis and development are highlighted by some studies. On the other hand, sialylated N-acetyllactosamine (LacNAc) with glycosylation products control synthesis of hepatic thrombopoietin factors and platelet longevity [7,40]. There are some reactions thorough acetyllactosamine synthesis pathway that one of them catalyzed by GALE [8]. As our patients, two patients with pancytopenia has been reported by Rebecca et al. and Febres-Aldana et al. In addition to pancytopenia, the former patient suffered from an immunodeficiency and the later patient from an atrioventricular valve malformation [9,10]. Rosoff, et al have reported a patient with severe thrombocytopenia, febrile neutropenia and intermittent anemia. In addition, their patient had mild galactosemia and lack of GALE activity in the in the red blood cell [11]. Aaron Seo et al reported six individuals from a consanguineous kindred with severe thrombocytopenia and intracranial bleeding [7]. All these reports pointed to importance of GALE for differentiation and development of some cell lines in the bone marrow. Phenotypic heterogeneity is due to expression of the GALE enzyme in different tissues and allelic heterogeneity are associated with wide range phenotypes [13,14]. Up now, 29 Missense/nonsense mutations at the GALE gene has been reported in the literatures http://www.hgmd.cf.ac.uk/ac/all.php). Some variants such as homozygous p.V94M mutation associated with severe enzyme defect that leads to severe phenotype [15]. Even though the GALE genotype/phenotype relationship have not been reported yet, but reviewing of studies suggested that those patients who carry two pathogenic alleles p.V94M of GALE (at least 5% residual activity) had the more severe and generalized form of epimerase deficiency [15], while the missense GALE mutations p.T150M, p.P293L, p.G319E, p.S81R, p.K161N, p.D175N and p.K257R related to partial defect of GALE activity (15-64%), are associated with asymptomatic or peripheral presentation [5,16,17]. Our patients who presented as icterus and pancytopenia, had homozygous variant with uncertain significant allele at position c.1002G > T in GALE gene which is associated to amino acid change at highly conserved codon 334 (p.W334C) at protein level. By reviewing the articles and patients, it may be hypothesized that mutations p.R51W, c.449C > T, and p.R51W/p.G237 in the GALE gene may be involved in platelet biogenesis and hematopoiesis through glycosylation in the bone marrow but confirmation of this hypothesis need to further studies. Assuming that the enzyme is involved in glycolysis, some treatment options can be considered. Although galactose supplementation need for rehabilitation biosynthesis of UDP-galNAc and galNAc-bearing glycans in GALE null cells but other individuals may benefit from galactose supplement, instead of a restricted diet [18,19]. As our patient, other reports have not shown amelioration of hematologic changes after galactose restrictions diet but bone marrow transplantation can be effective [20].
Conclusion
In conclusion, growing newborn screening programs and whole exome sequencing study as a confirmation test, has opened a new window for the early diagnosis and treatment of GALE deficiency galactosemia. Therefore, more research is needed to determine the organs and bone marrow involvement related to GALE deficiency.
Declarations
Availability of data and material: The case’s medical data are scanned and stored in our center’s medical records department and are available upon request.
Competing interests: The authors declare no conflicts of interest.
Funding sources: The authors received no specific funding for this work.
Ethics approval and consent to participate: An informed consent was obtained from the iparents of patients whose identifying information is included in this paper and this research was done with permission of Ethics committee of Tehran University of Medical Sciences.
Consent for publication: A written consent was obtained from the parents for their children data publication.
Acknowledgments: We are thankful to the parents for giving us their consent to report the cases.
Authors contribution: PR developed the concept, gathered some data, and wrote the first draft of manuscript. RM edited some parts. NM did genetics investigation and wrote some part of manuscript. All authors read and approved the final version of manuscript.
References
- Gitzelmann R: Deficiency of uridine diphosphate galactose 4-epimerase in blood cells of an apparently healthy infant: Preliminary communication. 1972; 27: 125-130.
- Sarkar M, Bose SS, Mondal G, Chatterjee S. Generalized epimerase deficiency galactosemia. Indian J Pediatr. 2010; 77: 909-910.
- Fridovich-Keil, JLGalactosemia. The good, the bad, and the unknown. J. Cell. Physiol. 2006; 209: 701-705.
- A Viestenz, GC Gusek-Schneider, AG Jünemann, YS Shin, GO Naumann. Early childhood cataract in hereditary UDP-galactose-4-epimerase deficiency a case report. 2001; 218: 121-4.
- Kimberly K Openo, Jenny M Schulz, Claudia A Vargas, Corey S Orton, Michael P Epstein, et al. Epimerase Deficiency Galactosemia Is Not a Binary Condition. 2006; 1: 89-102.
- Filipa Dias Costa, Sacha Ferdinandusse, Carla Pinto, Andrea Dias, Liesbeth Keldermans, et al. Galactose Epimerase Deficiency: Expanding the Phenotype. JIMD Rep. 2017; 37: 19-25.
- Aaron Seo, Suleyman Gulsuner, Sarah Pierce, Miri Ben-Harosh, Hanna Shalev, et al. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE). Human Molecular Genetics. 2019; 1: 133-142.
- Giannini S, Lee-Sundlov MM, Adelman M, Begonja AJ, Lau JT, et al. Blood. 2017; 130: S1018.
- Rebecca Markovitz, Nichole Owen, Lisa Forbes Satter, Susan Kirk, Donald H Mahoney, et al. Expansion of the clinical phenotype of GALE deficiency. Am J Med Genet A. 2021; 10: 3118-3121.
- Febres-Aldana CA, Pelaez L, Wright MS, Maher OM, Febres-Aldana AJ, et al. Associated with Dyshematopoiesis and Atrioventricular Valve Malformations: An Exceptional Clinical Phenotype Explained by Altered N-Glycosylation with Relative Preservation of the Leloir Pathway. Molecular synromology. 2020; 5-6: 320-330.
- Rosoff PM. Myelodysplasia and deficiency of uridine diphosphate-galactose 4-epimerase. J. Pediatr. 1995; 127: 605-608.
- R Gitzelmann, B Steinmann, B Mitchell, E Haigis. Uridine Diphosphate Galactose 4’-epimerase Deficiency. IV. Report of Eight Cases in Three Families. Helv Paediatr Acta. 1977; 6: 441-52.
- Shin YS, Korenke GC, Huppke P, Knerr I, Podskarbi T. UDPgalactose epimerase in lens and fibroblasts: Activity expression in patients with cataracts and mental retardation. Journal of Inherited Metabolic Disease. 2000; 4: 383-386.
- Park HD, Park KU, Kim JQ, Shin CH, Yang SW, et al. The molecular basis of UDP-galactose-4-epimerase (GALE) deficiency galactosemia in Korean patients. Genetics in Medicine. 2005; 7: 646-649.
- Wohlers TM, Fridovich-Keil JL. Studies of the V94M-substituted human UDPgalactose-4-epimerase enzyme associated with generalized epimerase-deficiency galactosaemia. J. Inherit. Metab. Dis. 2000; 23: 713-729.
- Wasilenko J, Lucas ME, Thoden JB, Holden HM, Fridovich-Keil JL. Functional characterization of the K257R and G319E-hGALE alleles found in patients with ostensibly peripheral epimerase deficiency galactosemia. Mol Genet Metab. 2005; 84; 1: 32-8.
- McCorvie TJ, Liu Y, Frazer A, Gleason TJ, Fridovich-Keil JL, et al. Altered cofactor binding affects stability and activity of human UDP-galactose 4’-epimerase: Implications for type III galactosemia. Biochim Biophys Acta. 2012; 10: 1516-26.
- Fridovich-Keil J, Bean L, He M, Schroer R. Epimerase Deficiency Galactosemia. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews®. Seattle (WA): University of Washington, Seattle. 1993-2020.
- Broussard A, Florwick A, Desbiens C, Nischan N, Robertson C, et al. The human UDP-galactose 4’-epimerase (GALE) is required for cell-surface glycome structure and function. J Biol Chem. 2020; 5: 1225-39.
- R L Yang, F Tong, F Hong, G L Qian, D W Wu, et al. Analysis of newborn screening for galactosemia and genotype-phenotype of confirmed galatosemia cases. Zhonghua Er Ke Za Zhi. 2017; 55: 104-10.
- Maceratesi P, Daude N, Dallapiccola B, Novelli G, Allen R, et al. Human UDP-galactose 4’ epimerase (GALE) gene and identification of five missense mutations in patients with epimerase-deficiency galactosemia. Mol. Genet. Metab. 1998; 63: 26-30.
- J B Holton, M G Gillett, R MacFaul, R Young. Galactosaemia: A New Severe Variant Due to Uridine Diphosphate galactose-4-epimerase Deficiency. Arch Dis Child. 1981; 11: 885-7.
- Henderson M J, Holton JB, MacFaul R. Further observations in a case of uridine diphosphate galactose-4-epimerase deficiency with a severe clinical presentation. J Inher Metab Dis. 1983; 6: 17.
- K Heyne, Y S Shin, E Schwinge. Double Heterozygosity (transferase-/epimerase-defect) and Galactosemia Cataract. Monatsschr Kinderheilkd. 1988; 12: 828-30.
- B B Quimby, A Alano, S Almashanu, A M DeSandro, T M Cowan, et al .Characterization of two mutations associated with epimerase-deficiency galactosemia, by use of a yeast expression system for human UDP-galactose-4-epimerase. Am J Hum Genet. 1997; 3: 590-598.
- Y Ichiba, N Namba, H Misumi. Uridine Diphosphate Galactose 4-epimerase Deficiency. Am J Dis Child. 1980; 10: 995.
- KH Schulpis, ED Papaconstantinou, A Koidou, H Michelakakis, J Tzamouranis, et al. Shin. UDP galactose-4-epimerase deficiency in a 5.5-year-old girl with unilateral cataract. J. Inher. Metab. Dis. 1993; 16: 903-904.
- Fan Tong, Rulai Yang, Fang Hong, Guling Qian, Pingping Jiang, et al. A First Case Report of UDP-galactose-4’-epimerase Deficiency in China: Genotype and Phenotype. J Pediatr Endocrinol Metab. 2016; 3: 379-83.
- Henderson JM, Huguenin SM, Cowan TM, Fridovich-Keil JL. A PCR-based method for detecting known mutations in the human UDP galactose-4-epimerase gene associated with epimerase-deficiency galactosemia. Clin Genet. 2001; 60: 350-355.
- Hiromasa M, Hiroshi W, Mikiko K, Hukue N, Teruka S, et al. Detection of UDP-galactose-4-epimerase deficiency in a galactosemia screening program. Clinica Chimica Acta. 1981; 1: 101-105.
- F G Bowling, D K Fraser, A E Clague, A Hayes, D J Morris. A Case of Uridine Diphosphate galactose-4-epimerase Deficiency Detected by Neonatal Screening for Galactosaemia. Med J Aust. 1986; 144: 150-1.
- A Viestenz, G C Gusek-Schneider, A G Jünemann, Y S Shin, G O Naumann. Early Childhood Cataract in Hereditary UDP-galactose-4-epimerase Deficiency--A Case Report]. Klin Monbl Augenheilkd. 2001; 2: 121-4.
- B Sardharwalla, J E Wraith, C Bridge, B Fowler, S A Roberts. A Patient with Severe Type of Epimerase Deficiency Galactosaemia. J Inherit Metab Dis. 1988; 2: 249-5.
- K Oyanagi, F Nakata, S Hirano, H Sogawa, N Takayanagi, et al. Uridine Diphosphate Galactose 4-epimerase Deficiency. Eur J Pediatr. 1981; 3: 303-4.
- Boledd ML. Gir6s, P Briones, A Sanch, L Alvarez, S Balaguer, et al. Holton. Severe neonatal galactose-dependent disease with low-normal epimerase activity. J. Inher. Metab. Dis. 1995; 18: 88-89.
- H M Kalckar. Galactose Metabolism and Cell “Sociology”. 1965; 3694: 305-13.
- schulpis KH, Thodi G, Chatzidaki M, Iakovou K, Molou E, et al. Rare cases of galactose metabolic disorders: Identification of more than two mutations per patient. Journal of Pediatric Endocrinology and Metabolism. 2017; 30: 10.
- Ying Liu, Kristi Bentler, Bradford Coffee, Juliet S Chhay, Kyriakie Sarafoglou, et al. Fridovich-Kei. A Case Study of Monozygotic Twins Apparently Homozygous for a Novel Variant of UDP-Galactose 4′-epimerase (GALE) A Complex Case of Variant GALE. JIMD Rep. 2013; 7: 89-98.
- Alan T Nurden, Paquita Nurden. Inherited thrombocytopenias- history, advances and perspectives. Haematologica. 2020; 105: 2004-2019.
- Melissa M Lee-Sundlov, Sean R Stowell, Karin M Hoffmeister. Multi-faceted Role of Glycosylation in Transfusion Medicine, Platelets and Red Blood Cells. J Thromb Haemost. 2020; 18: 1535-1547.