
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Cytogenetic profiling unveils Jacobs syndrome (47, XYY) - A unique chromosomal abnormality case from Nepal
Govardhan Joshi1,2,5*; Sandeep Thapa2,4; Santosh Khanal2,4; Pabitra Bista1,2; Anil Khadka2,5; Nikita Dhakal6; Ajay Jang Kunwar1,2,4; Nilam Thakur1,3,4,6
1Global Hospital and Education Foundation, India.
2Nova International Diagnostics, India, India.
3National Academy of Medical Sciences, India.
4Kathmandu Center for Genomics and Research Laboratory, India.
5Manmohan Memorial Institute of Health Sciences, India.
6Slavica IVF center, India.
*Corresponding Author : Govardhan Joshi
Global Hospital and Education Foundation, India.
Email: gobu2001@gmail.com
Received : Jan 20, 2024
Accepted : Feb 08, 2024
Published : Feb 15, 2024
Archived : www.jcimcr.org
Copyright : © Joshi G (2024).
Abstract
Jacobs syndrome (47, XYY) is a rare genetic condition observed in males due to incorrect segregation of chromosomes during the first or second meiotic cell division. A 38-year-old male with infertility issues, tall stature, and neurodevelopmental symptoms was diagnosed with a 47, XYY karyotype during a hospital visit prompted by his spouse’s adverse obstetric history. This syndrome depicts the impact on physical and cognitive development and underlines the relevance of providing genetic counseling and Assisted Reproductive Technology (ART) for couples with infertility. This study provides an understanding of XYY syndrome and highlights the need for ongoing research to enhance cytogenetic methodologies.
Keywords: Jacobs syndrome; ART; ASD; NIPS and karyotype.
Abbreviations: ART: Assisted Reproductive Technology; ASD: Autism Spectrum Disorder; NIPS: Non-Invasive Prenatal Screening; ECG: Electrocardiogram; ADHD: Attention Deficit Hyperactivity Disorder.
Citation: Joshi G, Thapa S, Khanal S, Bista P, Khadka A, et al. Cytogenetic profiling unveils Jacobs syndrome (47, XYY) - A unique chromosomal abnormality case from Nepal. J Clin Images Med Case Rep. 2024; 5(2): 2862.
Background
Jacob’s syndrome also recognized as XYY, is a rare genetic condition seen in about 1 out of 1000 male children that belongs to sex chromosome trisomies [1]. The syndrome remains undiagnosed in most of cases and often presents with a variable neurodevelopmental phenotype, incorporating developmental delays, cognitive impairments, and seizure [2,3]. While less prominent physical characteristics like tall stature, macroorchidism, macrocephaly and hypertelorism depict significant variability among affected individuals [4]. Additionally, infertility and Autistic Spectrum Disorders (ASD) are additional prevalent traits. Diagnosis of Jacob’s syndrome is often detained due to relatively subtle phenotypic changes with craniosynostosis, a fusion of cranial sutures, reported in approximately 1 in 2000 live births [5]. These individuals were diagnosed at a median age of 17.1 years, experiencing a substantial reduction in lifespan from 77.9 years (controls) to 67.5 years, coupled with an increase in total mortality rate compared to controls, with a hazard ratio of 3.6 (2.6-5.1) [6].
Jacob’s syndrome is typically non-inherited and arises during the father’s meiosis II. The manifestation of this syndrome in male offspring occurs when an extra Y chromosome is introduced to the sperm. The random nature of these events, predominantly paternal nondisjunction at meiosis II or occasionally postzygotic mitotic errors, emphasizes the importance of understanding the specific meiotic events contributing to 47, XYY syndrome [7].
The prenatal diagnosis of Jacob’s syndrome can be accomplished through invasive techniques such as amniocentesis or chorionic villus sampling, while Non-Invasive Prenatal Screening (NIPS) and microarray techniques are also available for screening purposes [8,9]. The application of multifarious interventions, spanning speech and occupational therapy, exhibits marked efficacy in mitigating challenges associated with XYY syndrome. The timely deployment assures accelerated issue resolution, while the integration of standard therapeutic modalities adeptly addresses additional aspects, such as acne and behavioral complexities, collectively fostering the improvement of well-being in affected individuals [10].
In this report, we present the case of an adult male who was diagnosed with XYY syndrome during a hospital visit, prompted by concerns related to his spouse’s adverse obstetric history. The patient’s medical examination, triggered by the challenging obstetric experiences of his spouse, led to the confirmation of the XYY karyotype, highlighting the importance of comprehensive genetic investigations in cases of reproductive health concerns.
Case presentation
A couple visited the Global Hospital and Education Foundation with complaints of bad obstetrics history. The couple has been married for the past 7 years, with a previous history of repeated miscarriages. The first miscarriage was spontaneous, whereas the second pregnancy loss was due to multiple congenital anomalies. The patient’s spouse is a 34-year-old woman with no major health issues on routine medical examination with a normal hormonal profile, and radiological findings like USG pelvis and hysterosalpingography examination revealed a normal uterus and patent fallopian tubes.
For the last five years, the couple faced difficulties in conceiving, experiencing two subsequent miscarriages. Despite her regular menstrual cycles, absence of dysmenorrhea or menorrhagia, no history of surgeries, and absence of sexually transmitted diseases, the couple revealed that the challenge of infertility was causing a considerable psychological burden in their lives.
The patient, a 38-year-old male with a height of 185.9 cm, a weight of 73 kg, and a BMI of 21.1 kg/m2 , exhibits a range of clinical features encompassing physical, neurodevelopmental, and psychosocial aspects. Notably, there was delayed puberty and abnormal testes size, suggesting potential hormonal or genetic factors contributing to infertility. The patient has ceased study due to learning disabilities along with motor coordination difficulties, hypotonia, and social difficulties. However, the absence of autism spectrum traits highlights a nuanced neurodevelopmental outline. There was synchronized arterial and ventricular activity on the ECG examination. Further, the patients’ semen analysis revealed normal semen volume (2 ml) color and viscosity, oligozoospermia with 4 million/ml, 40% motility, and 4% normal morphology. The basic laboratory analysis including hematological and biochemical tests, yielded results within normal limits. The elevated total testosterone level of 11.46 ng/ml, contributes to a broader understanding of hormonal dynamics in individuals with this rare sex chromosome aneuploidy.
Subsequently, the case was referred for chromosome analysis, and studies using the conventional cytogenetic technique unveiled a sex chromosomal abnormality. The presence of an abnormal non-mosiac 47, XYY karyotype was identified from the peripheral blood of the patient using the Geimsa Banding Technique (GTG), as depicted in Figure 1.
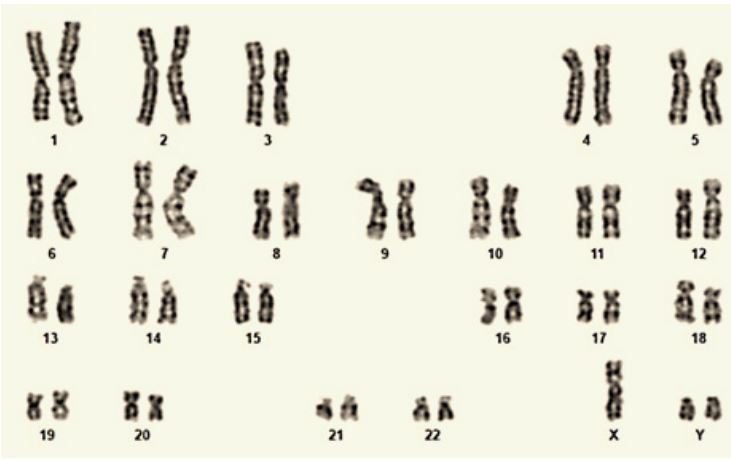
Table 1: Status of hematological and biochemical parameters.
| Parameter | Result |
Biological reference interval |
Unit |
|---|---|---|---|
| Hematological Parameters | |||
| Hemoglobin | 16.7 | 13-18 | g/dL |
| RBC | 4.96 | 4.2-5.4 | million/μL |
| PCV | 47.7 | 40-54 | % |
| Total WBC Count | 4700 | 4000-11000 | /μL |
| Neutrophils | 51 | 40-75 | % |
| Lymphocytes | 37 | 20-45 | % |
| Eosinophils | 4 | 1-6 | % |
| Monocytes | 8 | 2-10 | % |
| Basophils | 0 | <1 | % |
| Platelets | 146000 | 150000-450000 | /μL |
| MCV | 96.1 | 87±5 | fl |
| MCH | 33.8 | 27-33 | pg |
| MCHC | 35.2 | 30-35 | % |
| Renal Function Test | |||
| Urea | 27 | 15-45 | mg/dL |
| Creatinine | 1.0 | 0.9-1.3 | mg/dL |
| Sodium | 137 | 135-146 | meq/L |
| Potassium | 4.2 | 3.5-5.5 | meq/L |
| Liver Function Test | |||
| Total Bilirubin | 0.7 | 0.0-1.2 | mg/dL |
| Direct Bilirubin | 0.2 | 0.0-0.3 | mg/dL |
| SGPT (ALT) | 34 | 7-55 | IU/L |
| SGOT (AST) | 24 | 8-48 | IU/L |
| Alkaline Phosphatase | |||
| (ALP) | 154 | 70-380 | IU/L |
| Total Protein | 8.0 | 6-8 | gm/dL |
| Albumin | 4.2 | 3.5-5 | gm/dL |
| Thyroid Function Test | |||
| Free T4 | 1.72 | 0.78-2.19 | ng/dl |
| Free T3 | 6.32 | 2.77-5.27 | pg/ml |
| TSH | 1.85 | 0.46-4.66 | μIU/ml |
| Hormonal Assay | |||
| Total Testosterone | 11.46 | 1.7-7.8 | ng/ml |
| Growth Hormone | 0.04 | 0.020-3.893 | ng/ml |
Discussion
Jacobs syndrome is a rare condition associated with an additional Y chromosome in males that presents with mild symptoms with potential impact on physical and cognitive development [4]. The excess growth hormone and hyperthyroidism might be the causes of tall stature; however, these were adequate in this study. If the tall stature of a patient is attributed to an XYY karyotype, it is essential to assess their psychiatric status, fertility, and other non-specific symptoms [11].
Despite having a 47, XYY karyotype, many men demonstrate fertility, suggesting that the extra Y chromosome is frequently lost before meiosis, thus preserving their reproductive capabilities. Several studies comparing the sperm karyotypes of fertile and infertile XYY men reveal a prevalence of normal sperm. Besides, potential arrest points during spermatogenesis may result in maturation challenges and variable sperm concentrations [12,13]. Conversely, multiple studies underscore a notable occurrence of sperm mosaicism, aneuploidy, or hyperdiploidy in XYY men, emphasizing the potential risk of imparting abnormal genetics to offspring through the emergence of disomy YY cells during meiotic division [14].
During the diagnosis of Jacob syndrome, it is essential to differentiate it from Marfan syndrome a connective tissue disorder that also has tall stature along with cardiac abnormalities. Hence, a comprehensive cardiac activity assessment and genetic analysis of the fibirillin-1 gene mutation aid in differential diagnosis [15]. Similarly, the Sotos syndrome and Klinefelter syndrome have to be ruled out using NSD1 gene mutation and chromosomal studies respectively [16,17].
Genetic counseling is recommended for infertile couples, and assisted reproductive technology can be beneficial to those who have normal to oligozoospermia. It is critical to screen people with Jacobs syndrome who are prone have comorbid conditions such as asthma, tremors, seizure disorders, infertility, and psychological issues like Attention Deficit Hyperactivity Disorder (ADHD) and Autism Spectrum Disorder (ASD).
Conclusion
Overall, during reproductive health evaluations comprehensive genetic assessments are crucial in diagnosing Jacobs syndrome. The findings emphasize the diverse clinical manifestations, diagnostic challenges, and importance of genetic counseling and assisted reproductive technology. The advancements in cytogenetic methodologies are required to better understand and support individuals with this rare chromosomal abnormality.
Declarations
Acknowledgments: The authors express gratitude to patients for participating in this study. Special thanks to the laboratory staff for their technical expertise in conducting the cytogenetic analyses.
Funding: No funding was available.
Disclosure: No conflicts of interest were reported by the authors.
Ethics approval: Ethical committee approval not required.
Consent to participate: Written informed consent was obtained from the patient.
Consent for publication: Patient has been informed about publication of his case in a scientific journal for educational purpose.
References
- Van Rijn S. A review of neurocognitive functioning and risk for psychopathology in sex chromosome trisomy (47,XXY, 47,XXX, 47, XYY). Current Opinion in Psychiatry. 2019; 32.
- Wilson AC, King J, Bishop DVM. Autism and social anxiety in children with sex chromosome trisomies: an observational study. Welcome Open Res. 2019; 4.
- Zhang X, Liu X, Xi Q, Zhu H, Li L, Liu R, et al. Reproductive outcomes of 3 infertile males with XYY syndrome: Retrospective case series and literature review. Medicine. 2020; 99(9).
- Bardsley MZ, Kowal K, Levy C, Gosek A, Ayari N, Tartaglia N, et al. 47, XYY syndrome: clinical phenotype and timing of ascertainment. J Pediatr. 2013; 163(4): 1085-94.
- Swearingin TJ, Kirby BJ, Muzaffar AR. Presentation and Treatment of a Patient with Jacobs Syndrome and Metopic Craniosynostosis. Journal of Craniofacial Surgery. 2023; 34(7): 644-6.
- Stochholm K, Juul S, Gravholt CH. Diagnosis and mortality in 47, XYY persons: a registry study. Orphanet J Rare Dis. 2010; 5: 1-6.
- Bardsley MZ, Kowal K, Levy C, Gosek A, Ayari N, Tartaglia N, et al. 47, XYY syndrome: clinical phenotype and timing of ascertainment. J Pediatr. 2013; 163(4): 1085-94.
- Kypri E, Ioannides M, Touvana E, Neophytou I, Mina P, Velissariou V, et al. Non-invasive prenatal testing of fetal chromosomal aneuploidies: validation and clinical performance of the veracity test. Mol Cytogenet. 2019; 12: 1-7.
- Abedalthagafi M, Bawazeer S, Fawaz RI, Alajaji NM, Merrihew Heritage A, Faqeih EA. Non-Invasive Prenatal Testing: A Revolution Journey in Pre-Natal Testing. Front Med (Lausanne). 2023; 10: 1265090.
- Ross JL, Roeltgen DP, Kushner H, Zinn AR, Reiss A, Bardsley MZ, et al. Behavioral and social phenotypes in boys with 47, XYY syndrome or 47, XXY Klinefelter syndrome. Pediatrics. 2012; 129(4): 769-78.
- Jo WH, Jung MK, Kim KE, Chae HW, Kim DH, Kwon AR, et al. XYY syndrome: a 13-year-old boy with tall stature. Ann Pediatr Endocrinol Metab. 2015; 20(3): 170.
- Kim IW, Khadilkar AC, Ko EY, Sabanegh Jr ES. 47, XYY syndrome and male infertility. Rev Urol. 2013; 15(4): 188.
- Chatziparasidou A, Christoforidis N, Samolada G, Nijs M. Sperm aneuploidy in infertile male patients: a systematic review of the literature. Andrologia. 2015; 47(8): 847-60.
- Wong EC, Ferguson KA, Chow V, Ma S. Sperm aneuploidy and meiotic sex chromosome configurations in an infertile XYY male. Human reproduction. 2008; 23(2): 374-8.
- Yuan SM, Jing H. Marfan’s syndrome: an overview. Sao Paulo Medical Journal. 2010; 128: 360-6.
- Baujat G, Cormier-Daire V. Sotos syndrome. Orphanet J Rare Dis. 2007; 2(1): 1-6.
- Groth KA, Skakkebæk A, Høst C, Gravholt CH, Bojesen A. Klinefelter syndrome—a clinical update. J Clin Endocrinol Metab. 2013; 98(1): 20-30.