
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Bilateral vision loss with cranial nerve paralysis, revealing ischaemic pituitary apoplexy and acromegaly: A case report
Salma Abbas1,4*; Malak Riznat2,4; Mahjouba Boutarbouch1,4; Meriem Kajeou1,4; Hafsa EL Ouazzani3,4; Mohamed Anas Guerboub2,4; Nadia Cherradi3,4; Abdessamad El Ouahabi1,4
1Department of Neurosurgery, Specialties Hospital, Rabat, Morocco.
2Department of Endocrinology and Metabolic Diseases, Mohammed V Military Hospital, Rabat, Morocco.
3Department of Pathology, Specialties Hospital, Rabat, Morocco
4Mohammed V University of Medicine and Pharmacy of Rabat Morocco.
*Corresponding Author : Salma Abbas
Department of Neurosurgery, Specialties Hospital,
Rabat, Morocco
Email: Dr.s.abbas48@gmail.com
Received : Feb 12, 2024
Accepted : Feb 28, 2024
Published : Mar 06, 2024
Archived : www.jcimcr.org
Copyright : © Abbas S (2024).
Abstract
Pituitary Apoplexy (PA) is a potentially fatal clinical condition resulting from ischaemic or haemorrhagic infarction of the pituitary gland. The ischaemic subtype is even rarer and more severe. We present the case of a 26-year-old female patient who presented with headache, acute bilateral visual loss, fever, confusion, asthenia with classic features of acromegaly, and pupils with bilateral areactive mydriasis without light perception bilaterally with cranial nerve involvement. Magnetic Resonance Imaging (MRI) confirmed the presence of a pituitary macroadenoma, and endocrine investigations revealed panhypopituitarism. The patient underwent transsphenoidal evacuation and the diagnosis of ischaemic PA was confirmed by histology. Three months later, a follow-up MRI showed an empty bowel with remission of acromegaly, diabetes and mild unilateral visual recovery. Our case highlights the importance of considering ischaemic PA among the possible aetiologies of sudden cranial nerve deficits then early diagnosis and treatment are essential to improve outcome.
Keywords: Pituitary apoplexy; Panhypopituitarism; Magnetic resonance imaging; Macroadenoma; Pituitary ischemia; Acromegaly.
Citation: Abbas S, Riznat M, Boutarbouch M, Kajeou M, Ouazzani HEL, et al. Bilateral vision loss with cranial nerve paralysis, revealing ischaemic pituitary apoplexy and acromegaly: A case report. J Clin Images Med Case Rep. 2024; 5(3): 2903.
Introduction
Pituitary Apoplexy (PA) is a relatively rare but serious complication of pituitary adenoma. In most cases it is a medical and surgical emergency caused by an acute ischaemic infarction or haemorrhage in the pituitary gland. We report the case of a patient who was diagnosed with ischaemic PA, which may require rapid surgical decompression and a multidisciplinary approach involving endocrinologists, neuroradiologists and neurosurgeons.
Case presentation
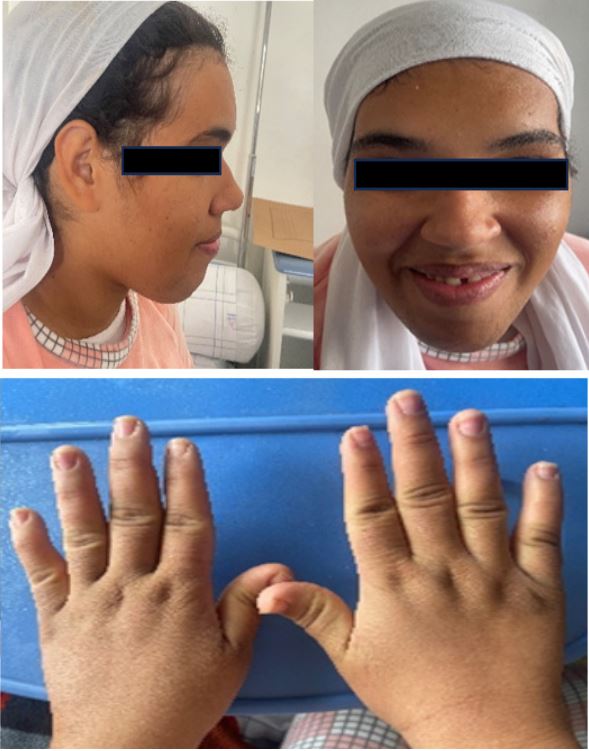
A 26-year-old male patient, single, laterally right-handed, with no particular medical history other than amenorrhoea for 3 years and an episode of frontoethmoidal sinusitis requiring several courses of antibiotics, presented with an acute decrease in bilateral visual acuity. The onset of symptoms occurred 5 days before admission, with the sudden onset of severe headache associated with jet vomiting, complicated by acute bilateral visual loss, all developing in the context of unquantified fever. On admission, the patient was confused with a GCS of 13/15 and deteriorating general condition, and haemodynamically stable with a blood pressure of 110/80 mmHg and temperature of 39°C. Clinical examination revealed an asthenic patient with classic acromegalic features (Figure 1), coarse facial features such as mandibular prognathism, rounded forehead, broad nose, thickened lips, large hands, large feet, macroglossia and hirsutism of the chin, and pupils with bilateral areactive mydriasis, without light perception on both sides, with a clear papilla on the right and pallor of the left papilla, in the fundus of the eye, with damage to the cranial nerves, namely bilateral paralysis of the VI, complete paralysis of the III on the right and partial on the left.
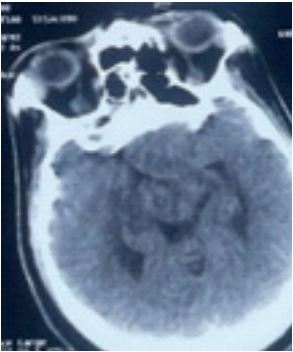
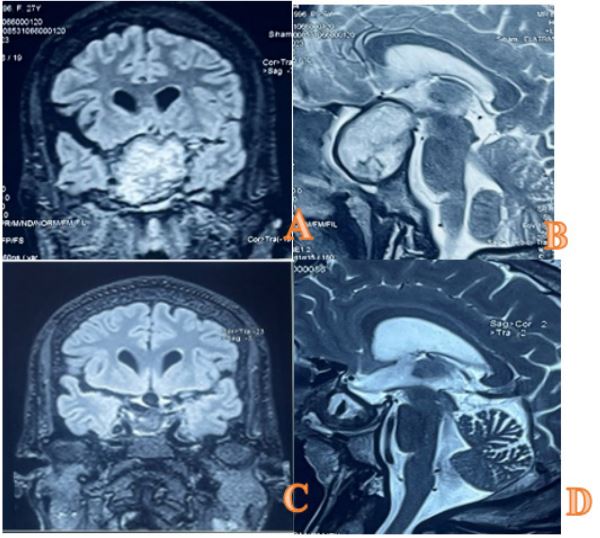
A brain MRI (Figure 3) was ordered and performed the following day, confirming the presence of a pituitary macroadenoma with hemorrhagic stigmata class KNOSP 3B measuring 32x41x42 mm apxtxh, T2 hypersignal, T1 iso signal with some areas of SWI asignal, peripherally enhanced.
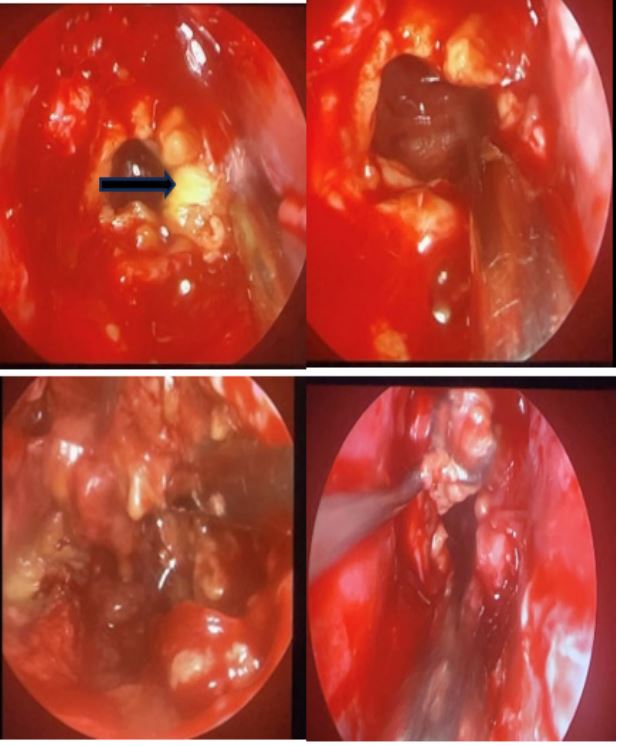
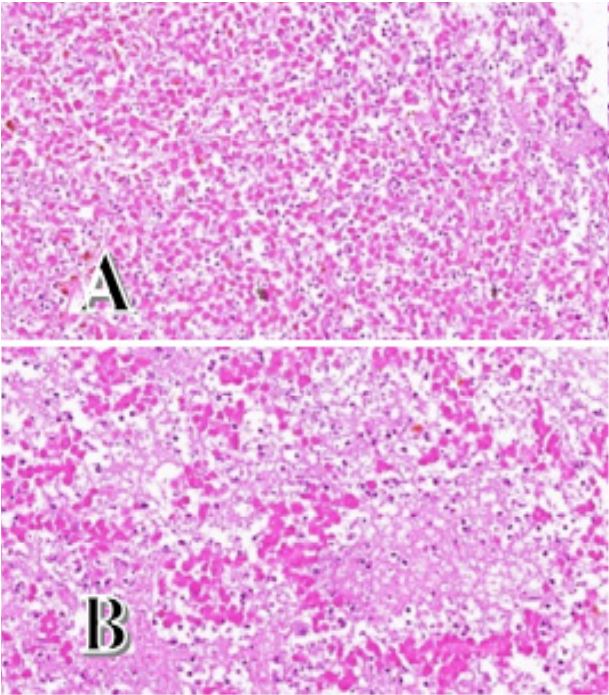
Anatomopathological examination revealed necrotic tissue with total fibro-inflammatory remodelling and no viable tumour cells (Figure 5). The necrosis was of an ischaemic type compatible with ischaemic PA.
The patient was immediately examined postoperatively for visual acuity and endocrine function. She presented with diabetes insipidus and was started on Minirin 2 cp/d in the morning. Hydrocortisone was started and HSHC 100 mg/6h given the high risk of corticotropic insufficiency. Her vision improved slightly on the right side, as did her immediate postoperative hormone levels (Table 2). The endocrinologist was consulted and therapy with levothyroxine and hydrocortisone was started with a three-month appointment, and the patient continued to follow up with both neurosurgery and endocrinology on an outpatient basis. A repeat MRI of the pituitary gland in 3 months showed a partially empty sellar (Figure 3). The sella turcica is occupied by material defined as post-operative changes related to possible accumulation of blood and scar tissue. Hormone levels were checked and he was found to have hormonal abnormalities as shown in Table 2.
Table 1: Grading system for pituitary apoplexy, Grade Clinical presentation.
| Grade | Clinical presentation |
|---|---|
| 1 | Asymptomatic |
| 2 | Endocrinological deficit only |
| 3 | Headache (acute onset or acute to chronic) |
| 4 |
Ophthalmoplegia due to
oculomotor paralysis of the
cranial nerves Ocular paresis (cranial nerves of the cavernous sinus) |
| 5 |
Visual disturbances or
reduced consciousness Visual
acuity or field deficit (or low Glasgow score excluding tests) |
Table 2: Comparative table of the different biological axes.
| Test | Before resection | After resection | After 3 months |
|---|---|---|---|
| PROLACTINE | <0,82 ng/ml | <0,82 ng/ml | <0,82 ng/ml |
| TSH | 0,080 UI/ml | 0,690 UI/ml | - |
| CORTISOL | <1 ug/dl | 7,3 ug/dl | 6,5 ug/dl |
| FSH | 1,20 mui/ml | 2,84 mui/ml | 4,61 mui/ml |
| LH | 0,12 mui/ml | 0,29 mui/ml | 2,06 mui/ml |
| TESTOSTERONE | 0,07 | - | 0,2 |
| GH | Error | 0,4 | - |
| IGF/1 | Error | 377 mg/ml | 270 mg/ml |
TSH: Thyroid-Stimulating Hormone; LH: Luteinizing Hormone; FSH: Follicle-Stimulating Hormone; GH: Growth Hormone; IGF-1 : Facteur De Croissance Analogue A L’insuline 1.
At the 3-month postoperative follow-up, our patient was admitted to the endocrinology unit for evaluation of endocrine and endocranial sequelae. She was conscious, well oriented in time and space, with a craniofacial dysmorphic syndrome typical of acromegaly, with an increase in the size of her extremities. Examination revealed blindness of the left eye, organomegaly dominated by a homogeneous goiter confirmed by ultrasound with the absence of digestive neoplastic lesions, hepatosplenomegaly and cardiomegaly confirmed by cardiac index. Skin and mucosal examination revealed hirsutism with a Ferrimen score of 23 and a frequency of facial shaving estimated at one in three days. The patient was also evaluated for other criteria for multiple endocrine neoplasia type 1 (MEN1), given her young age and the pituitary macroadenoma, which has returned without abnormality to date. Morphological evaluation of the disease revealed a vacant sella turcica with normal IGF1 levels 3 months postoperatively.
Discussion
The term apoplexy (derived from the Greek word apoplēxia, meaning “to be paralysed by a stroke”), first described by Pearce Baily in 1898, was first used by Brougham et al in 1950 after describing five cases of this clinical entity [1]. Pituitary Apoplexy (PA) is a rare and potentially fatal complication of neurosurgery. Its incidence ranges from 0.6% to 22.8% [10] and affects 2-12% of pituitary adenomas [2], 75-80% of whom have no known history of adenoma at the time of presentation [2]. Stroke in Growth Hormone (GH)-producing adenomas is a very rare event [12]; however, stroke due to ischaemic infarction from the tumor is very rare [10,15], with a higher incidence in men [4,13] and a mean age of presentation of 50-60 years [4]. The aetiology of PA is not well understood, but previous reports suggest that there are certain risk factors for PA, including a large tumour (macroadenoma) and reduced blood flow to the tumour; acute increase in blood flow to the pituitary or adenoma due to hypertension, diabetes, head trauma or pituitary irradiation; previous major surgery (especially orthopaedic and cardiac) or increased intracranial pressure; hormonal stimulation of the pituitary gland; and the use of anticoagulant drugs such as thrombolytic and antiplatelet therapy, dopamine or gonadotropin agonists, bromocriptine or oestrogens [2,3,7]. In 10-40% of cases, no predisposing factors can be identified [4].
The clinical symptomatology of PA is non-specific, usually sudden and severe retro-orbital or frontal headache, rapid visual disturbances with cranial nerve damage. According to the pituitary apoplexy classification system (Table 1), our patient is grade 5.
The endocrine deficit was directly attributable to the sudden and massive necrosis of the pituitary tissue, necessitating chronic hormone replacement therapy. Our patient presented with pan-hypopituitarism, in particular secondary adrenal insufficiency, hypothyroidism, Thyrotropin (TSH) deficiency, hypoprolactinemia and gonadotropin (LH and FSH) deficiency. She was started on hydrocortisone and levothyroxine. Diagnosis is usually made by MRI. Diffusion-Weighted Imaging (DWI) can detect the infarcted area shortly after a tumor infarction. The enhancement of the tumor border on MRI after gadolinium administration is a unique feature of ischaemic PA [15]. The 2010 UK PA management guidelines recommend that patients with severe neuro-ophthalmological deficits can be managed with early (<8 days) decompression surgery and immediate high-dose corticosteroid replacement [7,14]. The transsphenoidal approach (EET approach) is generally preferred for surgical treatment as it is the safest and fastest way to achieve adequate decompression of the optic chiasm and cranial nerves in patients with PA. As a result, morbidity and mortality are relatively low. At surgery, the tumor appeared as a yellow, cottage-cheese-like tissue with a soft texture and poor blood supply, different from that of a classic pituitary adenoma. Ischemic PA is defined by extensive areas of acellular necrosis, and immunohistochemical staining may or may not reveal the presence of residual neoplastic cells in these cases. However, they appear to be distinguished by the absence of residual neoplastic cells on immunohistochemical analysis described in some previous cases, which may be explained by the fact that extensive necrosis has destroyed all identifiable cells of the adenoma [5]. Phantom cells” are a typical manifestation characterised by phantom contours and absence of cellular structure [15]. Hormonal changes after surgery are very important in patients with pituitary adenoma as well as in PA. In our case, we found that all pituitary hormone levels were significantly begins to rise after surgery, except for prolactin and IGF1. Intra- or postoperative CSF leakage is the most common complication of the EET approach. Transient diabetes insipidus has been reported in approximately 2-20% of cases [13]. Our patient presented with diabetes insipidus controlled only after five days. The endocrine and neuro-ophthalmological prognosis of PA inevitably depends on the appropriateness of management during the acute and subacute phases of the disease. According to Ahmed Galal et al. the significant prognostic factor for visual recovery is the degree of preoperative visual deficit [6]. Recovery from cranial ocular neuropathy showed a higher rate of recovery when it was unilateral rather than bilateral. Pituitary hormone recovery was less favorable, with pituitary panhypopituitarism being a poor prognostic factor. Acromegaly is a multisystem disorder associated with metabolic, cardiac and musculoskeletal comorbidities that contribute to high mortality rates. Our patient presented with organomegaly including goiter, hepatosplenomegaly and cardiomegaly, secondary amenorrhoea and diabetes mellitus which resolved postoperatively. According to the latest consensus of November 2023, the diagnosis of acromegaly is confirmed by an IGF1 level >1.3, the upper limit of normal for age, in any patient with a suggestive clinical picture. The GH test still has its place, but only to assess the depth of the disease and therefore its prognosis [8,9].
Postoperatively, the normalisation of IGF1 3 months after resection of the neuroendocrine tumour is a criterion of remission and defines the success of surgery like in our case, Contrary to what was presented by Klimko et al. [8,9,11]. In 2020, a literature review of eleven cases published in the last 10 years describing the co-occurrence of acromegaly and pituitary apoplexy, where all patients were treated by transsphenoidal surgery. Six of the 11 patients still had elevated GH/IGF-1 levels postoperatively, which would be consistent with the literature reporting that the majority of GH-secreting pituitary adenomas remain functional after apoplexy.
Conclusion
Pituitary apoplexy is a diagnostic and therapeutic challenge that requires a multidisciplinary approach involving endocrinologists, neuroradiologists and neurosurgeons, with rapid treatment and standardised follow-up. Surgical decompression, indicated within 7-8 days of diagnosis, allows recovery of visual function and favourable outcomes. Hormonal deficiencies usually require medical replacement, and long-term follow-up is necessary for early detection of pituitary hormone deficiency and for detection of recurrence.
References
- Abdulbaki A Kanaan I. The impact of surgical timing on visual outcome in pituitary apoplexy: Literature review and case illustration.Surg Neurol Int. 2017; 6: 8. 16.doi: 10.4103/2152-7806.199557.
- Albani A, Ferraù F, Angileri F F, Esposito F, Granata F, Ferreri F, Cannavò S. Multidisciplinary Management of Pituitary Apoplexy. Intjendocrinol. 2016; 2016: 7951536. Doi: 10.1155/2016/7951536.
- Boellis A, Di Napoli A, Romano A, Bozzao A. Pituitary apoplexy: an update on clinical and imaging features. Insights Imaging. 2014; 5(6): 753-62. Doi: 10.1007/s13244-014-0362-0.
- Diane Donegan, Dana Erickson. Revisiting Pituitary Apoplexy. J Endocr Soc. 2022; 26;6(9): 113. Doi: 10.1210/jendso/bvac113.
- Funari A, Jeong SS, Pecorari IL, Flaquer I, Anderson CL, Agarwal V. Infarctive Apoplexy of Previously Healthy Pituitary Glands: A Small Case Series and Literature Review.J Neurol Surg Rep. 2023; 84(3): 71-79. Doi: 10.1055/s-0043-1770788.
- Galal A, El Farouk A O. Determinants of visual and endocrinological outcome after early endoscopic endonasal surgery for pituitary apoplexy. Surg Neurol Int. 2022; 13: 433. Doi :10.25259/SNI_642_2022.
- Geyik A, Durmaz M O, Dogan A, Ugur B K, Geyik S, İbrahim Erkutlu, Soner Yasar , Alparslan Kırık, Gulsah Kose, and Ali Nehir.Pituitary apoplexy: An emergent and potential life-threatening complication of pituitary adenomas Ulus Travma Acil Cerrahi Derg. 2022; 28(4): 483-489. Doi:10.14744/tjtes.2021.93539
- Giustina A, Biermasz N, Casanueva FF et al. Consensus sur les critères de diagnostic et de rémission de l’acromégalie. Pituitaire. 2023. Https://doi.org/10.1007/s11102-023-01360-1.
- Katznelson L, Laws ER Jr, Melmed S, Molitch ME, Murad MH, Utz A, Wass JA, Endocrine S (2014) Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014; 99: 3933-3951. Https://doi.org/10.1210/jc.2014-2700.
- Kim J P, Jin P B, Kim S B, Lim Y J. Pituitary Apoplexy due to Pituitary Adenoma Infarction. J Korean Neurosurg Soc. 2008; 43(5): 246-9. Doi: 10.3340/jkns.2008.43.5.246.
- Artsiom Klimko, Cristina Capatina. Pituitary Macroadenoma Presenting as Acromegaly and Subacute Pituitary Apoplexy: Case Report and Literature Review. Cureus. 2020; 12(8): 9612. Doi: 10.7759/cureus.9612.
- Mir S A,Masoodi S R,Bashir MI , Wani A I ,Farooqui K J , Kanth B ,Bhat A R . Dissociated hypopituitarism after spontaneous pituitary apoplexy in acromegaly. Indian Journal of Endocrinology and Metabolism 17(Suppl 1): S102-S104. 2013 Oct. DOI 10.4103/2230-8210.119518
- Siddharth Shah, Rumana Khan, Keenan Bayrakdar, et Christian Scott. Vision Deficit Due to Pituitary Apoplexy. Curéus. Mai 2023; 15(5): e38649.doi: 10.7759/cureus.38649.
- Xue-Qing Zheng, Xiang Zhou, Yong Yao, Kan Deng, Hui You, Lian Duan, Hui-Juan Zhu. Acromegaly complicated with fulminant pituitary apoplexy: clinical characteristic analysis and review of literature. Endocrine. 2023; 81(1): 160-167. Doi: 10.1007/s12020-023-03379-7. Epub 2023 May 17.
- Qiang Zhu, Yuchao Liang, Ziwen Fan, Yukun Liu, Chunyao Zhou, Hong Zhang, Tianshi Li, Yanpeng Zhou, Jianing Yang, Yinyan Wang, and Lei Wang. Ischemic Infarction of Pituitary Apoplexy: A Retrospective Study of 46 Cases From a Single Tertiary Center. Front Neurosci. 2021; 15: 808111. Doi: 10.3389/fnins.2021.808111.