
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
A rare case of acquired hereditary angioedema: Therapy response and outcome
Nonayra Bessa De Oliveira; Albertina Varandas Capelo*; Lívia Nascimento
Hospital Universitário Gaffrée e Guinle, Rio de Janeiro-RJ, Brazil.
*Corresponding Author : Varandas Capelo A
Hospital Universitário Gaffrée e Guinle, Rio de Janeiro-RJ, Brazil.
Email: albertinacapelo@hotmail.com
Received : Dec 30, 2024
Accepted : Jan 17, 2025
Published : Jan 24, 2025
Archived : www.jcimcr.org
Copyright : © Varandas Capelo A (2025).
Abstract
We present a case of Hereditary acquired angioedema in a young girl with Systemic Lupus Erythematosus and both deficiency and dysfunctional C1-INH. We also show prophylactic treatment burden.
Citation: De Oliveira NB, Varandas Capelo A, Nascimento L. A rare case of acquired hereditary angioedema: Therapy response and outcome. J Clin Images Med Case Rep. 2025; 6(1): 3439.
Introduction
Hereditary and acquired angioedema are two rare conditions that cause subcutaneous or submucosal edema associated with complement deficiencies, dysfunctions or other regulatory mechanisms [1,2]. While HAE results from a genetic mutation that affects the C1 inhibitor, Acquired Angioedema (AAE), which is ten times rarer than HAE, with a late onset, results due to the consumption of INH-C1 from malignant or autoimmune diseases, which may produce antibodies against INH-C1. So far, there is still little understanding and no licensed treatment for AAE [3-5]. So, we report a case of both HAE and AAE, the outcome and effectiveness of treatment.
We report the case of a 19-year-old black woman who suffered with recurrent, almost monthly episodes of angioedema on her face and limbs since fourteen years old. Some of these episodes were associated with joint pain, abdominal pain and vomiting. The patient reported a progressive increase in the frequency and intensity of symptoms over time. She denied comorbidities, medication use, other known triggers, or personal and family history of angioedema. She needed required emergency room treatment for several times, with spontaneous resolution in up to three days.
After dental treatment, the patient experienced an episode of severe facial angioedema, requiring emergency care. She underwent an evaluation with a rheumatologist, because of joint symptoms at that time, the clinical and laboratory results were not consistent with the diagnosis of Systemic Lupus Erythematosus (SLE). Later, she presented with a drop in CH50, C3, C4, and C1q, in addition to leukopenia, anemia, ANA 1:80 (fine dotted), positive anti-SSA, and positive lupus anticoagulant and vasculitis in the limbs, fever and arthralgia after a hard sun exposure, fever, and arthralgia in the knees and wrists, with a biopsy confirming SLE. Treatment for SLE was started with prednisone 20 mg, hydroxychloroquine 400 mg, and azathioprine 100 mg.
Nonetheless, the patient persisted with angioedema with no lupus exacerbation even with tranexamic acid (3 gr). We also tried danazol. It was not possible to measure anti-C1-INH antibodies. Tranexamic acid is well tolerated by most patients with AAE-C1-INH and is available in many countries but appear to be more effective in AAE-C1-INH than androgens [6].
One severe episode was treated with 3,000 units of human plasma C1-INH concentrate (pdC1-INH), intravenous tranexamic acid without any improvement. Then she was transfused with plasma, but developed generalized erythema and pruritus during the infusion. Some patients with AAE-C1-INH become less responsive to pdC1-INH over time, requiring higher doses to control symptoms [7,8]. The mechanism by which resistance to pdC1-INH develops appears to involve extremely rapid catabolism of the inhibitor protein, probably due to high levels of anti-C1-INH antibodies in serum.
Because of the persistent symptoms of angioedema without lupus worsening, it was suspected of HAE. The diagnostic evaluation with the immunologist revealed persistent decreased of C1-INH (3.9 mg/dL), and C1q (66.6 mg/dL), C4 (1 mg/dL) and C3 (70.2 mg/dL) and decrease of C1-INH function levels. Due to continuous attacks, we performed a genetic test that revealed a pathogenic variation in the SERPING1 gene, associated with hereditary angioedema with C1 inhibitor deficiency.
The diagnosis was confirmed as acquired hereditary angioedema with both deficiency and dysfunctional C1 Inhibitor. At this time, subcutaneous injections of 30 mg icatibant were indicated during the attacks, which provided complete and fast relief of symptoms. We also asked for lanadelumabe and c1 inhibitor human plasma C1-INH concentrate for prophylaxis but until now it was not possible.
The patient underwent uncomplicated dental surgery in a surgical center with tranexamic acid prophylaxis, without angioedema. However, after a confirmed pregnancy, she suffered a spontaneous pregnance loss and underwent curettage without complications. The SLE remained stable with the use of corticosteroids, hydroxychloroquine and azathioprine.
Despite this, she continued to experience frequent symptoms of angioedema. During three episodes of sudden facial swelling and difficulty breathing, she self-administered icatibant at home, reporting improvement in less than an hour (Figure 1).
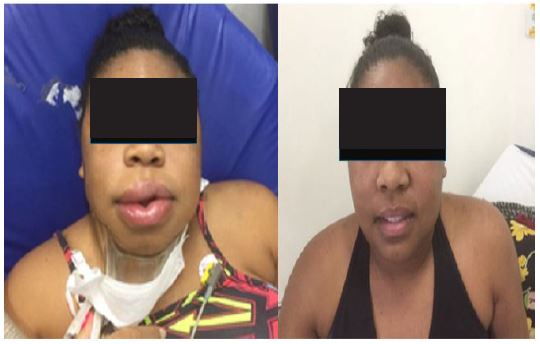
The patient got pregnant again and at this time she had already give birth to a healthy baby. During the pregnancy the patient suffered from a light angioedema event with spontaneous resolution in the first month. Recently, she suffered a face angioedema after a helmet hit her forehead. At this time she was treated with tranexamic acid. Therefore, this case illustrates the complexity of diagnosing and managing acquired hereditary angioedema, particularly in patients with concomitant SLE.
Treatment with icatibant, a bradykinin B2 receptor antagonist, has demonstrated efficacy in acute attacks of Hereditary and Acquired Angioedema (HAE), as evidenced by international studies and guidelines [9] including in Brazilian patients [10]. Icatibant therapy is especially relevant for the control symptoms, offering an important therapeutic alternative for these patients, especially in difficult-to-manage situations, such as the case described [9].
However, the second-line treatment was not effective and neither was a higher dose of human plasma C1-INH concentrate. Due to the side-effect of rituximab, the age of the patient, the pregnancy and the good control of SLE we did not consider Rituximab [11]. Plasma-derived C1 Inhibitor Concentrate (pdC1-INH) is used as an effective treatment for AAE attacks, although its use is off-label. Studies report that, despite its effectiveness in treatment, some failures may occur, possibly due to excessive consumption of C1-INH during attacks [10]. A cohort study conducted in Spain also observed the efficacy of pdC1-INH in angioedema attacks, with most patients treated with pdC1-INH or icatibant, both administered at home. However, in two cases there was an insufficient response to pdC1-INH. These findings suggest that, although the concentrate is generally effective, factors such as C1-INH consumption during attacks may influence the response to treatment, highlighting the importance of individually evaluating patients to optimize therapies [12,13].
Even when treating the underlying condition as in this case does not improve clinical course, we must consider based on few data published other prophylactic treatments according to the principles of the guidelines for the management of HAE [9].
There are still few data published about prophylactic treatments for acquired angioedema, particularly when both AAE and HAE coexist, relying on the principles of the guidelines for the management of HAE [9].
References
- Sinnathamby ES, Issa PP, Roberts L, Norwood H, Malone K, et al. Hereditary angioedema: Diagnosis, clinical implications, and pathophysiology. Advances in Therapy. 2023; 40(3): 814-27.
- Sohn BK, Fahrenholz JM, Awad JA, Fuchs HA. A case of acquired hereditary angioedema. The Journal of Allergy and Clinical Immunology: In Practice. 2020; 8(4): 1447-8.
- Cicardi M, Zingale LC, Pappalardo E, Folcioni A, Agostoni A. Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (Baltimore). 2003; 82: 274-81.
- Zanichelli A, Azin GM, Wu MA, Suffritti C, Maggioni L, et al. Diagnosis, course, and management of angioedema in patients with acquired C1-inhibitor deficiency. J Allergy Clin Immunol Pract. 2017; 5: 1307-13.
- Longhurst HJ, Zanichelli A, Caballero T, Bouillet L, Aberer W, et al. Comparing acquired angioedema with hereditary angioedema (types I/II): findings from the Icatibant Outcome Survey. Clin Exp Immunol. 2017; 188: 148-53.
- Tengborn L, Blombäck M, Berntorp E. Tranexamic acid: an old drug still going strong and making a revival. Thromb Res. 2015; 135: 231.
- Cicardi M, Zingale LC, Pappalardo E, et al. Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (Baltimore). 2003; 82: 274.
- Cicardi M, Bergamaschini L, Cugno M, et al. Pathogenetic and clinical aspects of C1 inhibitor deficiency. Immunobiology. 1998; 199: 366.
- Betschel S, Badiou J, Binkley K, Hébert J, Kanani A, et al. Canadian hereditary angioedema guideline. Allergy, Asthma & Clinical Immunology. 2014; 10: 1-18.
- Grumach AS, Henriques MT, Bardou MLD, Pontarolli DA, Botha J, et al. IOS Brazil Study Group. Icatibant use in Brazilian patients with Hereditary Angioedema (HAE) type 1 or 2 and HAE with normal C1-INH levels: findings from the Icatibant Outcome Survey Registry Study. Annals of Brazilian Dermatology. 2022; 97(4): 448-457.
- Buch MH, Smolen JS, Betteridge N, et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 2011; 70: 909-920.
- Agostoni A, Aygören-Pürsün E, Binkley KE, et al. Hereditary and acquired angioedema. The Journal of Allergy and Clinical Immunology. 2004; 114 (3): S51-S131.
- Baeza ML, Gonzalez-Quevedo T, Caballero T, Guilarte M, Lleonart R, et al. Angioedema due to acquired deficiency of C1-inhibitor: A cohort study in Spain and a comparison with other series. J Allergy Clin Immunol Pract. 2022; 10: 1020-8.