
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
Early Wiskott-Aldrich syndrome diagnosis and successful treatment in a 30-days-old male infant with congenital thrombocytopenia: Case report
Flora Tzifi1*; Marianna Tzanoudaki2; Marianna Varsou1; Christina Karastathi1; Afroditi Sakellaropoulou1; Aikaterini Kaisari3; Sofia Tantou2; Manolis Liatsis2; Evgenios Goussetis3; Anastasia Konidari1
1Second Department of Pediatrics, “P & A Kyriakou” Children’s Hospital, Athens, Greece.
2Department of Immunology & Histocompatibility, “Aghia Sophia” Children’s Hospital, Athens, Greece.
3Department of Hematopoietic Stem Cell Transplantation, “Aghia Sophia” Children’s Hospital, Athens, Greece.
*Corresponding Author : Flora Tzifi
Second Department of Pediatrics, “P & A Kyriakou”
Children’s Hospital, Thivon & Levadeias 11527,
Athens, Greece.
Tel: +030 2132009236; Email: fltzifi@med.uoa.gr
Received : Jan 04, 2025
Accepted : Jan 27, 2025
Published : Feb 03, 2025
Archived : www.jcimcr.org
Copyright : © Tzifi F (2025).
Abstract
Wiskott-Aldrich Syndrome (WAS) is a rare X-linked Inborn Error of Immunity (IEI), characterized by micro-thrombocytopenia, severe eczema, recurrent life-threatening infections, and tendency to malignancies and autoimmunity. Diagnosis during the neonatal period, in the absence of family history, is not only challenging, but also crucial for Hematopoietic Stem Cell Transplantation (HSCT) success and overall survival. We describe the case of a one-month-old male infant with congenital thrombocytopenia and bloody stools, initially misdiagnosed as alloimmune neonatal thrombocytopenia. Clinical picture and the implementation of flow cytometry led to prompt diagnosis of WAS. The patient was successfully treated with HSCT. Differential diagnosis of congenital thrombocytopenia is complex and multifactorial. Patients with severe WAS can display symptoms on the first day of life. Clinical picture, assessment of simple diagnostic tools such as blood cell count/ peripheral blood smear and flow cytometry can lead to correct diagnosis and successful treatment. Finally, we highlight the importance of training pediatricians in IEI reference centers and continuous education on IEI, providing better treatment results and quality of life in WAS patients.
Keywords: Wiskott-Aldrich syndrome; Infants; Congenital thrombocytopenia; Transplantation.
Abbreviations: WAS: Wiskott-Aldrich Syndrome; HSCT: Hematopoietic Stem Cell Transplantation; IEI: Inborn Error of Immunity; CMV: Cytomegalovirus; IVIG: Intravenous Gamma Globulin; WAS-P: Wiskott-Aldrich Syndrome Protein.
Citation: Tzifi F, Tzanoudaki M, Varsou M, Karastathi C, Sakellaropoulou A, et al. Early Wiskott-Aldrich syndrome diagnosis and successful treatment in a 30-days-old male infant with congenital thrombocytopenia: Case report. J Clin Images Med Case Rep. 2025; 6(2): 3450.
Introduction
Wiskott-Aldrich Syndrome (WAS) is a rare X-linked Inborn Error of Immunity (IEI), caused by mutations in the WAS gene located on the X chromosome (Xp11.22-p11.23). WAS is presented with a wide disease spectrum; in severe cases it is characterized by micro-thrombocytopenia, difficult to treat eczema, haematochezia, recurrent infections, increased malignancy risk, and autoimmunity phenomena later in life [1,2]. Median age of diagnosis occurs usually during late infancy, when the complete phenotype is unraveled, whilst diagnosis is more common in the three first months of life in patients with a positive family history [3]. Herein, the case of a one-month-old male infant with congenital thrombocytopenia and without family history for inherited disorders is presented; prompt diagnosis was achieved due to high clinical suspicion, careful assessment of blood cell counts and implementation of flow cytometry measurement of Wiskott Aldrich Protein (WAS-P). The patient was successfully treated with Hematopoietic Stem Cell Transplantation (HSCT) from a matched unrelated donor.
Case presentation
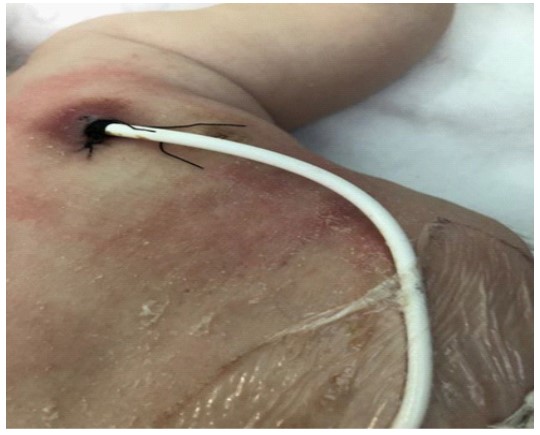
A one-month-old male infant was admitted via the emergency department for investigation of congenital thrombocytopenia. He was the first offspring of a non-consanguineous Caucasian couple, born in a neighboring country. Mother was referred to be infected with SARS-COV-2 and Cytomegalovirus (CMV) during the third trimester of pregnancy. On admission, the patient had thrombocytopenia, as revealed from blood cell count, mild facial eczema and occasionally bloody stools. Family history was negative for inherited or other disorders. The neonate presented with fever and thrombocytopenia on the first day of life. In the neonatal care unit, he had been treated with antibiotics and platelet transfusions. He had been discharged with normal platelet count, but three days later (at the age of 20-days-old) he was readmitted to the neonatal care unit due to bloody stools and thrombocytopenia (33.000/mm3). He received further platelet transfusion and gamma globulin (IVIG) and was referred for further evaluation by hematologist and gastroenterologist. On admission, the patient had normal vital signs and satisfactory growth. Clinical examination revealed mild facial eczema, seborrheic dermatitis, and single palmar line. Initial blood cell count evaluation displayed eosinophilia and thrombocytopenia (20.000/mm3) (Table 1). Infection investigation revealed positive IgG antibodies for CMV and positive urine CMV PCR. Fundoscopy, cardiologic evaluation, ultrasound of brain and abdomen were normal. X-ray of the right arm was not suggestive of Tar syndrome. On admission, the age of the patient, the absence of family history for genetic disorders, the possible asymptomatic infection with CMV, the good general state and growth of the infant, the mild eczema and the occasionally bloody stools could lead to misdiagnosis or the delay of correct diagnosis. Differential diagnosis of congenital thrombocytopenia included several etiological factors such as infectious agents (viral or bacterial, eg CMV), alloimmune thrombocytopenia, hematological diseases (congenital leukemia), genetic syndromes (trisomy 21,13,18), autoimmune and metabolic disorders and hereditary thrombocytopenia. Careful assessment of blood cell count revealed that platelets displayed low MPV (5fl), but a) it was not certain whether this finding was reliable and b) micro-thrombocytopenia on blood smear was not certain. The combination of a possible micro-thrombocytopenia with eczema and colitis, led to targeted immunologic evaluation which revealed marginal low IgM serum levels and CD8+ lymphopenia (Table 1). Assessment of WAS-protein expression was performed as soon as possible, on first week of hospitalization. WAS-P was undetectable by flow cytometry, indicating severe WAS phenotype (Figure 1). Subsequent HLA analy- sis was performed, in order to find HLA compatible donor for HSCT awaiting for genetic confirmation of diagnosis, targeting to one gene (WAS gene) and not to whole exome sequencing. Sequence analysis and deletion/duplication of the WAS gene revealed hemizygous mutation c.631C>T (pArg211) at exon 7, which could be accounted for the absence of WAS protein [3]. This is a rare mutation that has been described in few patients with severe WAS [3]. Severe WAS leads to progressive immunodeficiency, and patients are prone to severe infections, cancer and autoimmunity, conditions that shorten dramatically life expectancy. The patient received supportive treatment for almost six months, remaining mainly hospitalized in our clinic before his transfer to the HSCT care unit. Supportive treatment included periodic infusions of intravenous gamma-globulin, chemoprophylaxis, and rationalization of platelet transfusions. The infant presented with oral petechiae and gradually severe dysphagia due to deep painful oral ulcers. He also displayed severe diapers dermatitis that partially responded to a short course cortisone treatment. Eczema also spread to the rest of the body, mimicking the clinical picture of atopic dermatitis. Skin dermatitis, local bleeding, and infection at the Hickman catheter entry point, prevented catheter stabilization [4] (Figure 2). At the age of seven months old, the patient received peripheral blood stem cell transplant from fully matched unrelated adult donor, following a myeloablative conditioning regimen. Engraftment was achieved on day 18 post -transplantation, with subsequent platelet rise (>20.000/mm3) and neutrophil rise > 500/mm3) on day 41+. The early post-transplant period was uneventful, apart from acute GVHD grade II reaction, which responded to steroids. The patient had full donor chimerism and received periodic gamma globulin infusions and cyclosporine. He also received treatment for mild CMV infection. At discharge from the transplantation department he received immunosuppressive agents, corticosteroids for two months and cyclosporine for twelve months. The infant currently two and a half-year old, is asymptomatic, with a normal blood cell count. Follow-up assessment of the patient is performed annually by the transplantation department as well as from our clinic. No adverse or unanticipated events occurred.
Discussion
In the case presented, thrombocytopenia was accidentally revealed on the first day of life due to a possible neonatal bacterial infection, but immunological evaluation was not considered even when the neonate presented with colitis and thrombocytopenia. Differential diagnosis of congenital thrombocytopenia is complex and includes numerous etiological factors, infectious, genetic, hematological, and hereditary. Diagnosis was established very early, at the age of one month old, mainly to careful assessment of a) clinical picture of the patient and b) the availability of quick WAS-P assessment by flow cytometry. Avoidance of unnecessary laboratory investigation and proper management of the patient were established [5-7]. Transplantation was performed, before immune deterioration led to op- portunistic infections with subsequent worst outcome. It should be noted that the median age of diagnosis of WAS is reported to be 12 months old [1,2,3,5], while onset of symptoms is usually at 3 months of age in severe cases. Early diagnosis at the age of one month old is extremely rare and usually is referred in cases with family history. Furthermore, initial presentation may be not as specific as to warrant referral to an immunologist, unless there is increased awareness for IEIs. Finally, since WAS cannot be readily detected by the neonatal IEI screening programs [8], which are currently performed in a limited number of countries worldwide, training general pediatricians in IEI reference centers and continuous education on immunodeficiencies is of utmost importance.
Table 1: Blood cell count and immunologic evaluation of the patient at his admission.
| Blood cell count | Reference values | Igs | Values | Age matched normal range | Lymphocyte subpopulations | % | Absolute numbers | Normal ranges | |
|---|---|---|---|---|---|---|---|---|---|
| White Blood cells | 18300/mm³ | 6000-17500 | IgA | 31 mg/dl | 6-81 mg/dl | CD3+ | 48.4% | 3630 | 2500-5600 |
| Neutrophils | 39.2% | 40-75 | IgM | 31 mg/dl | 31-135 mg/dl | CD4 | 36.7% | ||
| Lymphocytes | 35.9% | 20-45 | IgG | 761 mg/dl | 224-1072 mg/dl | CD8+ | 17% | ||
| Monocytes | 15.7% | 2-10 | IgE | 8.42 | <1.94 | CD3+CD8+ | 8.3% | 622/µl | 590-1600 |
| Eosinophiles | 7.6% | 0-4 | CD3+CD4+ | 36.1% | 2708/µl | 1800-4000 | |||
| Baseophiles | 1.6% | 0-1 | CD19+ | 27.6% | 2070/µl | 430-3000 | |||
| Band neutrophils | 11% | CD4+CD45RO+ | 6.4% | ||||||
| Hb (g/dl) | 10.7 mg/dl | 10-18 | CD4CD45RA+ | 25.7% | |||||
| Hct% | 32.2 | 31-55 | CD16+/CD56+ | 18.5% | 1388/µl | 170-1400 | |||
| MCV (fl) | 95.3 | 85-123 | HLA-DR+ | 30.8% | |||||
| MCH (fl) | 31.7 | 28-38 | NKT | 2.3% | |||||
| PLTs | 27000/mm³ | 130.000-400.000 | CD4+/CD8+ | 2.1% | |||||
| MPV | 5 fl |

References
- Candotti F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J Clin Immunol. 2018; 38: 13-27.
- Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: A comprehensive review. Ann N Y Acad Sci. 2013; 1285: 26-43.
- Gulácsy V, Freiberger T, Shcherbina A, Pac M, Chernyshova L, Avcin T, et al. Genetic characteristics of eighty-seven patients with the Wiskott-Aldrich syndrome. Mol Immunol. 2011; 48: 788-92.
- S Ray, R Stacey, M Imrie, J Filshie. A review of 560 Hickman catheter insertions. Anaesthesia. 1996; 51: 981-5.
- Suri D, Rikhi R, Jindal AK, Marcolina M, Barzaghi F, et al. Wiskott Aldrich Syndrome: A Multi-Institutional Experience From India. Front Immunol. 2021; 16: 12: 627651.
- Gunnink SF, Vlug R, Fijnvandraat K, van der Bom JG, Stanworth SJ, et al. Neonatal thrombocytopenia: etiology, management and outcome. Expert Rev Hematol. 2014; 7: 387-95.
- Rivers E, Worth A, Thrasher AJ, Burns SO. How I manage patients with Wiskott Aldrich syndrome. Br J Haematol. 2019; 185: 647-655.
- Giżewska M, Durda K, Winter T, Ostrowska I, Ołtarzewski M, et al. Newborn Screening for SCID and Other Severe Primary Immunodeficiency in the Polish-German Transborder Area: Experience from the First 14 Months of Collaboration. Front Immunol. 2020; 16: 11:1948.