
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
Sporadic opto-chiasmatic glioma in a child: A case report
Basma Beqqali, MD1*; Izi Zineb, MD1; Kabila Badr, MD1; Oubaddi Tlait, MD1; Mesbah Oumaima, MD1; Allali Nazik, PhD1; El Haddad Siham, PhD2; Chat Latifa, PhD1
1Department of Mother-Child Imaging, Ibn Sina University Hospital Center, Morocco.
2Department of Radiology, Ibn Sina University Hospital Center, Morocco.
*Corresponding Author : Basma Beqqali, MD
Department of Mother-Child Imaging, Ibn Sina
University Hospital Center, Morocco.
Tel: +212667799987
Email: beqqali.basma@gmail.com
Received : Jan 13, 2025
Accepted : Feb 10, 2025
Published : Feb 17, 2025
Archived : www.jcimcr.org
Copyright : © Beqqali B (2025).
Abstract
Optic nerve glioma is a rare, slow-growing brain tumor primarily affecting children, often associated with Neurofibromatosis type 1 (NF1). This case report presents a 6-year-old girl diagnosed with unilateral optic nerve glioma, emphasizing her clinical presentation, diagnostic process, and management approach. The patient initially presented with progressive vision impairment and left eye proptosis, prompting further investigation. MRI confirmed a mass confined to the optic nerve, with characteristic imaging features supporting the diagnosis. A multidisciplinary team of pediatric neurologists, oncologists, and ophthalmologists evaluated treatment options, ultimately recommending chemotherapy to preserve vision and control tumor growth. This report underscores the importance of early detection and intervention in optic nerve gliomas to improve visual outcomes and quality of life. Through this case, we highlight the clinical and imaging findings that can aid in timely diagnosis and discuss the role of tailored treatment strategies in managing these tumors effectively.
Keywords: Optic nerve glioma; Pediatric; Neurofibromatosis type 1; MRI; Vision impairment.
Citation: Beqqali B. Sporadic opto-chiasmatic glioma in a child: A case report. J Clin Images Med Case Rep. 2025; 6(2): 3471.
Introduction
Optic Pathway Gliomas (OPGs) are rare pediatric brain tumors, accounting for about 1-5% of all childhood brain tumors, and often present in children under the age of 10. They arise primarily within the optic nerve and chiasm and are frequently associated with Neurofibromatosis type 1 (NF1), with an estimated incidence of 20-40% in NF1 patients [1,2]. These tumors are classified as low-grade astrocytomas, typically exhibiting slow growth, which often allows for excellent overall survival. However, OPGs vary greatly in presentation, ranging from asymptomatic to aggressively symptomatic. Key symptoms include progressive vision loss, proptosis, and changes in the optic disc, which can significantly affect the child’s quality of life, depending on the tumor’s location and progression [1].
While generally benign, the impact of optic pathway gliomas on vision and potential neurological complications means that early diagnosis and prompt management are critical. Treatment for OPGs is complex, requiring a multidisciplinary team to tailor the approach based on tumor size, location, associated symptoms, and progression rate. This case report describes the clinical journey of a 6-year-old girl diagnosed with an optic nerve glioma, illustrating the importance of coordinated care in the management of pediatric OPGs.
Case report
A 6-year-old girl was brought to the ophthalmology clinic with progressive vision loss and proptosis in her left eye over several months, accompanied by intermittent headaches and nausea. Family history was negative for genetic disorders or tumors. Examination revealed decreased visual acuity in her left eye, a relative afferent pupillary defect, and optic disc swelling on fundoscopy. MRI showed a large mass centered on the optic chiasm, extending into the hypothalamus and adjacent structures, resulting in hydrocephalus and signs of increased intracranial pressure.
A multidisciplinary team of pediatric oncologists, neurologists, and ophthalmologists convened to determine treatment, considering the tumor’s location and visual function. They initiated chemotherapy with vincristine and carboplatin, aiming to control growth and preserve vision, with regular follow-up imaging to monitor response.
Discussion
Optic Pathway Gliomas (OPGs) exhibit varied clinical behaviors, from asymptomatic to locally aggressive, depending on tumor size, location, and NF1 association. Most are low-grade with favorable survival [1]. NF1-related OPGs often progress indolently, sometimes stabilizing or regressing without intervention, unlike sporadic cases, which require more active management [2]. Studies show NF1-related OPGs mainly affect the orbital nerve (66%) and chiasm (62%), whereas sporadic cases typically involve the chiasm (91%) [3].
Symptom presentation varies; common signs include vision loss and proptosis in larger tumors, while hypothalamic involvement can lead to polyuria, obesity, and endocrine issues due to hypothalamic-pituitary compression [4,5]. Tumors reaching significant size, as in this case, may raise intracranial pressure, causing headaches, neurological deficits, and hydrocephalus.
MRI is critical for diagnosing and monitoring OPGs. Thin-slice MRI helps evaluate optic nerve enlargement and tumor characteristics across sequences. On T1-weighted images, OPGs are typically hypo- to isointense, while T2-weighted images show iso- to hyperintensity, with variable contrast enhancement [6]. Sporadic OPGs more often display cystic components and pronounced gadolinium enhancement compared to NF1-related gliomas and are likelier to affect the posterior optic pathway [5], as observed here.
Dodge et al.’s anatomical classification categorizes these tumors by location: optic nerves (stage 1), chiasm (stage 2), and hypothalamus or nearby structures (stage 3) [7], aiding prognosis and treatment planning, with posterior or hypothalamic involvement generally linked to poorer outcomes.
Differential diagnosis for OPG includes optic nerve meningioma, distinguishable by the lack of calcification present in optic nerve sheath meningioma. When tumors involve the chiasm, consider other lesions, including pituitary region masses.
Management of pediatric OPGs, based on SIOP-E-BTG and GPOH guidelines, is individualized by age, tumor location, and progression. Observation is typical in asymptomatic NF1-related cases. However, chemotherapy is initiated for progressive vision loss or vital structure involvement. Surgery aims for complete resection but is limited due to vision loss risk. Standard chemotherapy includes vincristine and carboplatin, with vinblastine and bevacizumab as alternatives. Radiotherapy is generally avoided in NF1 patients due to the risk of radiation-induced malignancies [8]. Targeted therapies, like MAPK inhibitors, show promise but require more research [8].
Prognosis for most OPGs is favorable, with low malignant transformation risk and a 20-year survival rate over 90% for children older than three. Long-term visual impairment remains concerning, with poor prognostic factors including early onset, postchiasmatic location, hypothalamic symptoms, intracranial pressure, incomplete resection, and lack of NF1 mutations. NF1-related OPGs generally present before the chiasm, carrying a better prognosis, while tumors at or beyond the chiasm exhibit higher progression and mortality rates [9,10].
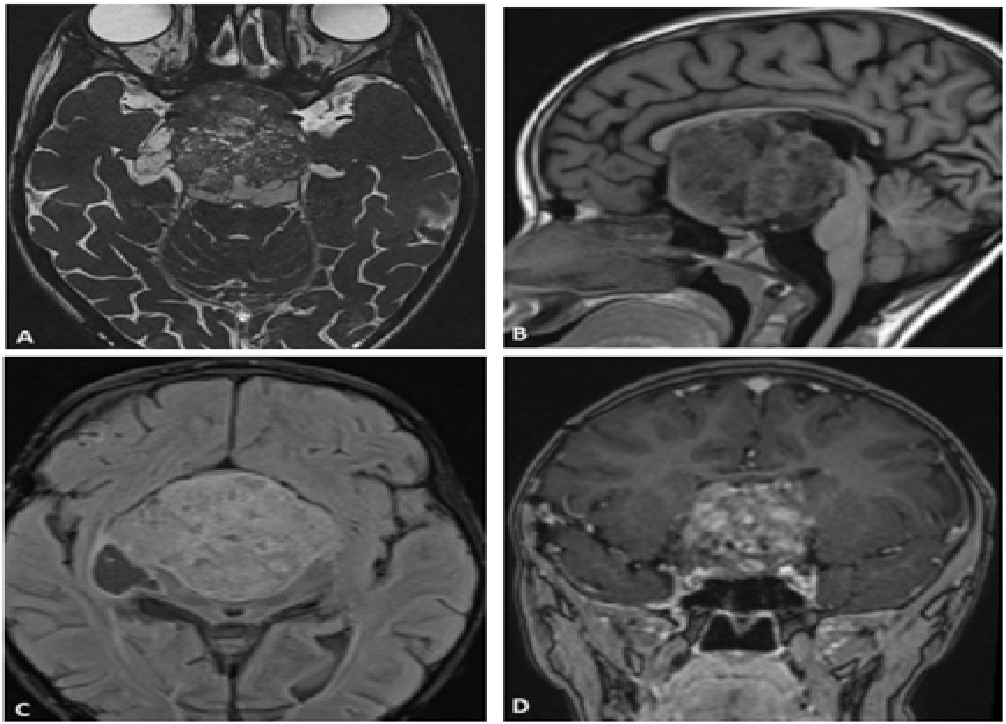
Conclusion
Despite two centuries of study, Optic Pathway Gliomas (OPGs) remain challenging to manage. Accurate radiological measurements are crucial for evaluating treatments and correlating visual and radiological outcomes, especially with the advent of new targeted therapies.
Declarations
Conflict of interest: The author(s) declare that they have no conflicts of interest that could have inappropriately influenced them in the writing of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval: No ethical approval was required for this article
Informed consent: Written informed consent was obtained from the patient for the anonymized information to be published in this article.
Guarantor: Beqqali Basma.
References
- Lafay-Cousin L, Holm S, Qaddoumi I, Nicolin G, Bartels U, Tabori U, et al. Weekly vinblastine in pediatric lowgrade glioma patients with carboplatin allergic reaction. Cancer. 2005; 103: 2636–2642.
- Shamji MF, Benoit BG. Syndromic and sporadic pediatric optic pathway gliomas: review of clinical and histopathological differences and treatment implications. Neurosurg Focus. 2007; 23: E3.
- Millar WS, Tartaglino LM, Sergott RC, Friedman DP, Flanders AE. MR of malignant optic glioma of adulthood. American journal of neuroradiology. 1995; 16: 1673-6.
- Huang M, Patel J, Patel BC. Optic Nerve Glioma. Available online: https://pubmed.ncbi.nlm.nih.gov/32491801/.
- Avery RA, Fisher MJ, Liu GT. Optic Pathway Gliomas. J. Neuro-Ophthalmol. 2011; 31: 269–278.
- Fangusaro J, Witt O, Hernáiz Driever P, et al. Response assessment in paediatric low-grade glioma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020; 21: e305–e316.
- Taylor T, Jaspan T, Milano G, Gregson R, Parker T, Ritzmann T, et al. PLAN Study Group. Radiological classification of optic pathway gliomas: experience of a modified functional classification system. Br J Radiol. 2008; 81: 761-6.
- Modrzejewska M, Olejnik-Wojciechowska J, Roszyk A, Szychot E, Konczak TD, Szemitko M, et al. Optic Pathway Gliomas in Pediatric Population-Current Approach in Diagnosis and Management: Literature Review. J Clin Med. 2023; 12: 6709.
- Hata J, Barbour M, Huang MA. LGG-02. Pediatric Low-Grade Glioma Risk Stratification in the Molecular Era. Neuro-Oncology. 2021; 23: i31.
- Robert-Boire V, Rosca L, Samson Y, Ospina LH, Perreault S. Clinical Presentation and Outcome of Patients with Optic Pathway Glioma. Pediatr. Neurol. 2017; 75: 55–60.