
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
Anatomical casting diagnosis of a fetus with interrupted aortic arch combined with ventricular septal defect: A case report and literature review
Haidong Ai1#; Hongping Ou1#; Yanli Huang3#; Yongmei Jia1#; Feng Li4#; Chuang Guo1#; Chaoyi Lv1#; Yu Wang1,2,5*
1Department of Ultrasound Medicine, Xiangyang Central Hospital, Hubei University of Arts and Science, Xiangyang, Hubei 441021, China
2The Institute of Maternal-Fetal Integrated Intelligent Diagnostics, Xiangyang Central Hospital, Hubei University of Arts and Science, Xiangyang, Hubei 441021, China
3Department of Obstetrics and Gynecology, Xiangyang Central Hospital, Hubei University of Arts and Science, Xiangyang, Hubei 441021, China.
4Radiographic Imaging Department, Xiangyang Central Hospital, Hubei University of Arts and Science, Xiangyang, Hubei 441021, China
5Department of Ultrasound, The Second Affiliated Hospital of Nanhua University, Hengyang, Hunan 421001, China
#Contributed equally.
*Corresponding Author : Yu Wang
The Institute of Maternal-Fetal Integrated
Intelligent Diagnostics, Xiangyang Central Hospital,
Affiliated Hospital of Hubei University of Arts and
Science, 136 Jingzhou Street, 441020, Xiangyang,
Hubei, China.
Email: 287383672@qq.com.
Received : Feb 14, 2025
Accepted : Mar 17, 2025
Published : Mar 24, 2025
Archived : www.jcimcr.org
Copyright : © Wang Y (2025).
Abstract
Interrupted aortic arch (IAA) is a relatively rare congenital vascular malformation, with an incidence of less than 1% among congenital heart diseases (CHD). This condition is classified as a ductdependent anomaly, wherein blood from the left ventricle fails to flow adequately through the descending aorta, compelling reliance on the right ventricle for perfusion to the lower extremities via the ductus arteriosus. If the ductus arteriosus closes after birth, the infant may rapidly develop heart failure and perish. Fetal echocardiography serves as the primary diagnostic modality for such conditions; however, it is beset by certain limitations, particularly in fully visualizing vascular variations and abnormal branching patterns. Interrupted aortic arch frequently coexists with other complex cardiovascular anomalies, and while echocardiography offers significant advantages in visualizing intracardiac structures, it presents challenges in classifying these conditions. Vascular casting techniques provide a comprehensive depiction of the integrity and three-dimensionality of extricated fetal vessels, facilitating in-depth observation of vascular pathways, morphology, and positional relationships, thus offering an intuitive representation of local and overall anatomical interrelations. This article reports a case of a fetal interrupted aortic arch accompanied by a ventricular septal defect, aiming to provide valuable insights for future clinical practice.
Keywords: Anatomical casting; Interrupted aortic arch; Fetal congenital heart disease.
Citation: Haidong A, Hongping O, Yanli H, Yongmei J, Wang Y, et al. Anatomical casting diagnosis of a fetus with interrupted aortic arch combined with ventricular septal defect: A case report and literature review. J Clin Images Med Case Rep. 2025; 6(3): 3521.
Case presentation
A 26-year-old primigravida with no significant medical history denied any familial occurrences of congenital heart disease or genetic disorders. Down syndrome screening yielded a lowrisk result, and there was no history of exposure to radioactive toxins during the early stages of pregnancy. Following induction, the anatomical casting of the fetus revealed that the heart predominantly occupied the left thoracic cavity, with the apex oriented downward to the left. The left atrial appendage appeared elongated and finger-like, whereas the right atrial appendage was broad and triangular. A casting agent was observed traversing the upper portion of the interventricular septum, indicating a ventricular septal defect (Figures 1C, 1D). The aorta originated from the left ventricle with a notably narrowed diameter, while the pulmonary artery emerged from the right ventricle with a significantly widened diameter. The aorta was positioned posteriorly and to the left of the pulmonary artery, which lay anteriorly and to the right, resulting in a crossing relationship at their origins. The ascending aorta and aortic arch exhibited marked constriction, with the aortic arch deviating slightly to the left of the midline. The arch sequentially gave rise to the right brachiocephalic artery, the left common carotid artery, and the left subclavian artery before being abruptly interrupted, continuing as the descending aorta, indicative of an interrupted aortic arch (Figure 1E, 1F). Additionally, the diameters of the main pulmonary artery and bilateral pulmonary arteries were notably enlarged. The ductus arteriosus was located to the left of the aorta, exhibiting an increased diameter and extending posteriorly to the descending aorta. The left brachiocephalic vein coursed above the aortic arch, transversely draining into the superior vena cava, which then emptied into the right atrium. Casting diagnosis: complex fetal congenital heart disease; 1. Interrupted aortic arch (Type A); 2. Ventricular septal defect. The casting specimens are illustrated in Figures 1A-1F.
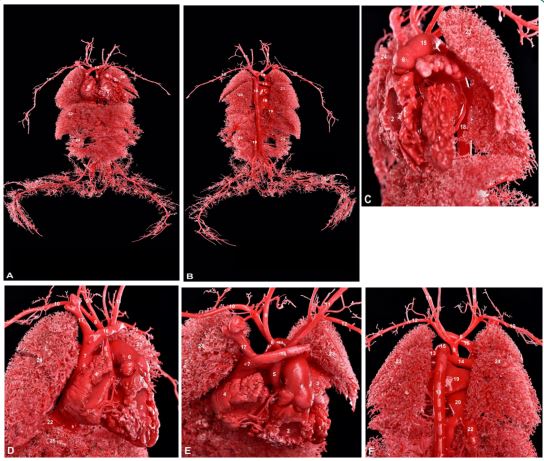
A: Overall frontal view of the mold;
B: Overall posterior view. 1 left ventricle, 2 right ventricle, 3 left atrial appendage, 4 right atrial appendage, 5 aorta, 6 pulmonary artery, 7 superior vena cava, 8 brachiocephalic artery, 9 right common carotid artery, 10 right subclavian artery, 11 left common carotid artery, 12 left subclavian artery, 13 left pulmonary artery, 14 left brachiocephalic vein, 15 right brachiocephalic vein, 16 descending aorta, 17 left atrium, 18 right atrium, 19 inferior vena cava, 20 left lung, 21 right lung, 22 liver, 23 left kidney, 24 right kidney.
C: Left lateral view of the thorax;
D: Right lateral perspective;
E: Superior view of the heart;
F: Posterior view of the heart. 1 left ventricle, 2 right ventricle, 3 left atrial appendage, 4 right atrial appendage, 5 aorta, 6 pulmonary artery, 7 superior vena cava, 8 brachiocephalic artery, 9 right common carotid artery, 10 right subclavian artery, 11 left common carotid artery, 12 left subclavian artery, 13 left pulmonary artery, 14 right pulmonary artery, 15 ductus arteriosus, 16 left brachiocephalic vein, 17 right brachiocephalic vein, 18 descending aorta, 19 left atrium, 20 right atrium, 21 pulmonary veins, 22 inferior vena cava, 23 left lung, 24 right lung, 25 liver.
Discussion/conclusion
Interrupted aortic arch (IAA) is a rare congenital cardiovascular malformation characterized by the discontinuity of either the ascending aorta or the aortic arch, accounting for approximately 1% to 3% of congenital heart disease (CHD) cases [1]. IAA is classified into three distinct types based on the site of interruption: Type A occurs distal to the left subclavian artery; Type B is situated between the left subclavian artery and the left common carotid artery; and Type C lies between the brachiocephalic artery and the left common carotid artery [2]. Notably, the highest incidence of interruption occurs between the left subclavian artery and the left common carotid artery, representing about two-thirds of all cases, while the occurrence between the brachiocephalic artery and the left common carotid artery is the least common, comprising less than 5% [3].
This condition must be differentiated from severe coarctation of the aorta (CoA), which presents as a focal eccentric narrowing of the aortic segment, typically located distal to the left subclavian artery near the junction of the ductus arteriosus. However, in severe cases of CoA, complete loss of luminal continuity may occur, leading to hemodynamic changes that closely resemble those of IAA. While arterial contrast-enhanced imaging can effectively distinguish between the two, its teratogenic effects on the fetus render it unsuitable for routine prenatal screening. Consequently, echocardiography remains the most crucial diagnostic tool for identifying congenital vascular anomalies. Nonetheless, the limitations of two-dimensional echocardiographic imaging can hinder the accurate assessment of complex vascular malformations and their spatial relationships with adjacent structures. Additionally, the hemodynamic alterations in Type A IAA and severe CoA are often quite similar, sometimes making differentiation challenging [4].
We report a case of Type A IAA in which post-delivery casting revealed significant constriction of the ascending aorta and aortic arch, with the arch positioned slightly to the left of the midline. Sequentially emanating from the arch was the right brachiocephalic artery, left common carotid artery, and left subclavian artery, before discontinuing the descending aorta, indicating Type A aortic arch interruption (Figures 1A, 1B, 1E, 1F). The left atrial appendage appeared elongated and finger-like while casting material was observed traversing the upper portion of the ventricular septum, suggesting a ventricular septal defect. The aorta originated from the left ventricle with a narrowed internal diameter, whereas the pulmonary artery arose from the right ventricle with a markedly widened internal diameter (Figures 1C, 1D). This case demonstrated not only IAA but also coarctation of the aorta and a ventricular septal defect, which are challenging to distinguish through echocardiography alone. Our casting method effectively elucidates the major cardiac vessels’ origins, pathways, and variations, thereby enriching the understanding of the complex anatomical structures and spatial relationships inherent in large vascular anomalies. This provides critical insights for future clinical practice and accurate fetal cardiac ultrasound diagnoses.
Declarations
Authors’ contributions: Y.W. planned the concept of the study; F.L. and C.G. performed the data processing and analysis. Experiments were conducted, analyzed, and interpreted by C.L.; H.A. drafted the manuscript; H.O., Y.H., and Y.J. edited the manuscript and provided valuable suggestions. All authors contributed to the article and approved the submitted version.
Competing interests: The authors declare that they have no competing interests.
Funding: This work was supported by grants from the Xiangyang R&D Plan Project (2022YL44A); China International Medical Foundation (Z-2014-08-2309-30); the Clinical Research Center for Fetal Complex Malformations, Hubei Provincial Department of Science and Technology (2021LC002); the Key Project of the Medical and Health Technology Plan, Xiangyang Science and Technology Bureau, Xiangyang City, China (2021YL19); the General Project Guidance Program, Hubei Provincial Department of Science and Technology (2022CFC042).
References
- Hanneman K, Newman B, Chan F. Congenital Variants and Anomalies of the Aortic Arch. Radiographics. 2017; 37(1): 32-51.
- CELORIA GC, PATTON RB. Congenital absence of the aortic arch. Am Heart J. 1959; 58: 407-13.
- Landeras LA, Chung JH. Congenital Thoracic Aortic Disease. Radiol Clin North Am. 2019; 57(1): 113-125.
- Vriend JW, Lam J, Mulder BJ. Complete aortic arch obstruction: interruption or aortic coarctation. Int J Cardiovasc Imaging. 2004; 20(5): 393-6.