
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
A case of neonatal encephalopathy with non-epileptic myoclonus mimicking a congenital CMV-infection. How could two different pathologies result in a similar MRI phenotype?
Adriana Cristofano1; Valentino Perrotta2; Camilla Russo1*; Eugenio Maria Covelli1
1Neuroradiology Unit, Department of Pediatric Neurosciences, Santobono-Pausilipon Children’s Hospital, 80129 Naples, Italy.
2Neonatology and Neonatal Intensive Care Unit, Santobono-Pausilipon Children’s Hospital, 80129 Naples, Italy.
*Corresponding Author : Camilla Russo
Neuroradiology Unit, Department of Pediatric Neurosciences, Santobono-Pausilipon Children’s Hospital, 80129 Naples, Italy.
Tel: 0039 081 2205682;
Email: camilla_russo@hotmail.it
Received : Mar 19, 2025
Accepted : Apr 08, 2025
Published : Apr 15, 2025
Archived : www.jcimcr.org
Copyright : © Russo C (2025).
Abstract
KCNQ2-related disorders are rare autosomal dominant channelopathies caused by mutations in KCNQ2, a gene that encodes a subunit of the voltage-gated potassium channel Kv7, resulting in variable phenotypes. Neonatal encephalopathy with non-epileptic myoclonus is one of the less common and more complex phenotypes of KCNQ2 channelopathies. It is caused by a gain-of-function variant of KCNQ2 and is characterized by a distinctive clinical-electrographic expression but nonspecific brain MRI findings. Here, we report the case of a newborn diagnosed with neonatal encephalopathy with non-epileptic myoclonus, whose MRI phenotype mimics congenital CMV infection.
Keywords: Congenital CMV-infection; KCNQ2-related disorders; Neonatal encephalopathy with non-epileptic myoclonus; Magnetic resonance imaging.
Abbreviations: EEG: Electroencephalogram; MRI: Magnetic Resonance Imaging; PVPC: Periventricular Pseudocyst; KCNQ2: Potassium Voltage-Gated Channel Subfamily Q Member 2.
Citation: Russo C, Cristofano A, Perrotta V, Maria Covelli E. A case of neonatal encephalopathy with non-epileptic myoclonus mimicking a congenital CMV-infection. How could two different pathologies result in a similar MRI phenotype?. J Clin Images Med Case Rep. 2025; 6(4): 3554.
Introduction
Potassium voltage-gated channel subfamily Q member 2 (KCNQ2)-related disorders are rare autosomal dominant channelopathies caused by mutation in KCNQ2, a gene that encodes a subunit of the voltage-gated potassium-channel Kv7. Such mutations result in variable phenotypes, ranging from benign seizures in infancy to developmental and epileptic encephalopathy [1]. Neonatal encephalopathy with non-epileptic myoclonus is one of the less common and more complex manifestation of KCNQ2-channelopaties, caused by a gain-of-function variant of KCNQ2 instead of the other more frequent loss-of-function variants [1]. In affected children, clinical features are profound encephalopathy with hypotonia evident from birth, respiratory impairment, and severe developmental delay. Myoclonus can be spontaneous or triggered by sound or touch [1]. Electroencephalogram (EEG) recording is peculiar, showing an invariant burst-suppression pattern without electrical cortical signs during jerking movements, while multifocal epileptiform discharges could be recorded later in life [1]. At the onset no abnormality can be detected at brain magnetic resonance imaging (MRI) [1], while a variable degree of brain atrophy and hypomyelination can be observed in children who develop seizures and a severe neurodevelopmental impairment, regardless of genotype [1]. Herein, we describe the clinical manifestations and the brain MRI findings of a patient with neonatal encephalopathy with non-epileptic myoclonus Mimicking A Congenital Cytomegalovirus (CMV)-infection.
Case study
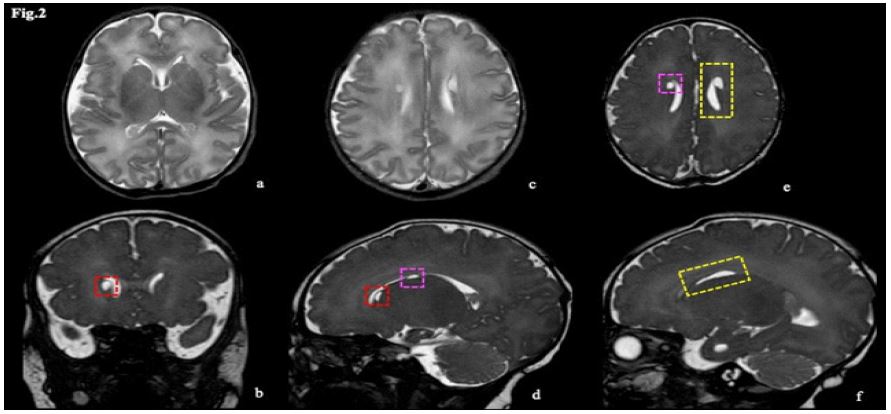
The patient was a female newborn with a gestational age of 38+0/7 weeks, the first child of non-consanguineous parents. The pregnancy was uneventful, and delivery occurred in the setting of an altered cardiotocographic tracing, where a possible role of antenatal seizures could not be retrospectively ruled out. APGAR scores at the 1st and 5th minutes were 9 and 9, respectively. During her first week of life, she was referred to our institution due to uncontainable jerks. Neurological examination revealed axial hypotonia, a strong alert response, and both spontaneous and sound/touch-triggered myoclonus. Episodes of unstable breathing requiring oxygen therapy were also reported. Continuous video EEG showed a severely abnormal background with a suppression-burst pattern, myoclonus, and hiccups without EEG correlation, and irregular or periodic breathing on polygraphy. Simultaneously with the diagnostic workup, pyridoxine and pyridoxal 5-phosphate were administered, leading to an apparent attenuation of symptoms. Brain MRI revealed an edematous appearance of the parenchyma due to diffuse T2 hyperintensity of the white matter and mild cerebral swelling, along with bilateral and nearly symmetric periventricular pseudocysts (PVPCs) localized in both the frontal and temporal areas, temporopolar cystic lesions, and focal anterior enlargement of the temporal horns of the lateral ventricles. These findings primarily suggested a congenital infection during the late gestational trimester, most likely CMV (Figures 1,2). However, CMV and all other congenital infections were subsequently ruled out. Biochemical screening and metabolic analysis were unremarkable. Finally, t-NGS analysis identified a heterozygous variant in the KCNQ2 gene (NM_172107.4: c.597C>A; p. Ser199Arg). Consequently, neonatal encephalopathy with non-epileptic myoclonus was established as the final diagnosis.

Discussion & conclusions
Neonatal encephalopathy with non-epileptic myoclonus is one of the less common and more complex phenotypes of KCNQ2-channelopaties, caused by a gain-of-function variant of KCNQ2 and, as in the other KCNQ2-related disorders, brain MRI have never revealed any peculiar findings at onset. In this patient, brain MRI findings consist of PVPCs, localized both in frontal and in temporal areas, temporopolar cystic lesions and focal anterior enlargement of temporal horns of lateral ventricles; brain parenchyma appears lightly edematous, too. Gathering these findings, a CMV congenital infection is hypothesized as the most likely diagnosis. CMV is the most common cause of perinatal viral infection, affecting 0.2-2.2% of all neonates [2]. The specific CMV neurotrophic effect, together with inflammatory neurotoxic factors and placenta impairment, may compromise neuronal proliferation, incomplete neuronal migration, and cortex organization, ending up in abnormal brain morphogenesis and maturation [3,4]. When the infection occurs during the late gestational trimester, the neuroimaging phenotype could be less severe, consisting of polar/anterior temporal lesions, PVPCs, calcifications, slightly ventriculomegaly, intraventricular septa or white matter lesions, spotted as areas of increased T2-weighted signal intensity [5]. These MRI findings could be differently and variably arranged therefore the same specific MRI pattern is not always present in all affected newborns [3]. Concerning PVPCs, it is known that their formation is due to prenatal lysis of undifferentiated germinal matrix cells, that they could be considered as a mark of impaired neuronal migration and that the prognosis is poor when they are not isolated, especially if other findings occur [6]. Depending on how other clinical and imaging data are combined, anterior temporal cystic lesions could suggest congenital muscular dystrophy, genetic leukodystrophies, Aicardi-Goutières syndrome or congenital CMV-infection [7]. Even though T2-hypersignal of white-matter abnormalities, PVPCs and temporopolar lesions could be a common MRI pattern among a wide spectrum of different pathologies, congenital CMV-infection should be the first hypothesis to rule out in the diagnostic algorithm. The overlapping MRI phenotypes could be explained both by the recent evidence of dysregulated expression of 4029 genes in CMV-infected human cerebral organoids [8], including KCNQ2-gene, and by the emerging role of KCNQ2 in neurodevelopment since antenatal period [9-11], as confirmed by the association between KCNQ2 mutations and mild alterations in cortical development, as described at a microscopic level in two newborns neuropathology reports [12,13]. However, more studies are needed to deeply understand the physiopathology behind congenital infections and neonatal onset channelopathies.
Declarations
Funding: No intramural and/or extramural funding was used to support this work.
Conflict of interest: The authors declare that there is no conflict of interests regarding the publication of this paper.
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent: Informed consent was obtained from all individual participants included in the study, as part of the standard of care.
Authors contribution: All authors make substantial contributions to conception and design, and/or acquisition of data, and/or analysis and interpretation of data according to ICMJE recommendations. All those who have made substantive contributions to the article have been named as authors. The case has already been presented as an e-poster to the 1st European Pediatric Neuroradiology Congress 2024.
References
- Miceli F, Soldovieri MV, Weckhuysen S, Cooper E and Taglialatela M. KCNQ2-Related Disorders. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. 2010. https://www.ncbi.nlm.nih.gov/books/NBK32534/.
- Ciobanu AM, Gica N, Gica C, Botezatu R, Furtuna M, et al. Cytomegalovirus Infection in Pregnancy - Counselling Challenges in the Setting of Generalised Testing.Maedica (Bucur). 2020; 15: 253-257.
- Doneda C, Parazzini C, Righini A, Rustico M, Tassis B, et al. Early cerebral lesions in cytomegalovirus infection: Prenatal MR imaging.Radiology. 2010; 255: 613-621.
- Vande Walle C, Maris F, Schiettecatte E and Herregods N. The value of magnetic resonance imaging in congenital cytomegalovirusinfection: A systematic review. Pediatr Radiol. 2024; 54: 2157-2174.
- Triulzi F, Doned C and Parazzini C. Neuroimaging of pediatric brain infections. Expert Rev Anti Infect Ther. 2011; 9: 737-751.
- Sun C, Zhang X, Chen X, Wei X, Chen Y, Yang A et al. Evaluation of MRI Features and Neurodevelopmental Outcomes for Prenatally Diagnosed Periventricular Pseudocysts. Front Pediatr. 1999; 9: 68.
- Nunes RH, Pacheco FT, da Rocha AJ. Magnetic resonance imaging of anterior temporal lobe cysts in children: Discriminating special imaging features in a particular group of diseases. Neuroradiology. 2014; 56: 569-577.
- Egilmezer E, Hamilton ST, Foster CSP, Marschall M, Rawlinson WD. Human cytomegalovirus (CMV) dysregulates neurodevelopmental pathways in cerebral organoids. Commun Biol. 2024; 7: 340-352.
- Swayne LA, and Wicki-Stordeur L. Ion channels in postnatal neurogenesis: potential targets for brain repair. Channels (Austin). 2012; 6: 69-74.
- Dirkx N, Miceli F, Taglialatela M and Weckhuysen S. The Role of Kv7.2 in Neurodevelopment: Insights and Gaps in Our Understanding. Front Physiol. 2020; 11: 570588.
- Smith RS and Walsh CA. Ion Channel Functions in Early Brain Development. Trends Neurosci. 2020; 43:103-114.
- Legros L, Adle-Biassette H, Dozières-Puyravel B, Khung S, Elmaleh-Bergès M, et al. Neuropathology findings in KCNQ2 neonatal epileptic encephalopathy. Seizures. 2022; 99: 36-39.
- Dalen Meurs-van der Schoor C, van Weissenbruch M, van Kempen M, Bugiani M, Aronica E, Ronner H et al. Severe Neonatal Epileptic Encephalopathy and KCNQ2 Mutation: Neuropathological Substrate. Front Pediatr. 2014; 2: 136-142.