
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 6
Familial clustering in chronic hypersensitivity pneumonitis: Diagnostic dilemmas and role of lung biopsy
Syed Murtaza H Kazmi1*; Wajahat Mirza2; Muhammad Sajeel Turab3; Ahmad Talha Tariq4; Sundus Dadan5; Zafar Ali6; Rashed Nazir7
1Chief of Department and Consultant, Department of Pulmonology, Shifa International Hospital, Islamabad, Pakistan.
2Fourth Year MBBS Student, Shifa College of Medicine, Shifa Tameer-e-Millat University, Islamabad, Pakistan.
3Fifth Year MBBS Student, Shifa College of Medicine, Shifa Tameer-e-Millat University, Islamabad, Pakistan.
4Resident, Department of Internal Medicine, Shifa International Hospital, Islamabad, Pakistan.
5Research Associate, Shifa’s Clinical Research Centre, Shifa International Hospital, Islamabad, Pakistan.
6Section Head and Consultant, Department of Histopathology, Shifa International Hospital, Islamabad, Pakistan.
7Consultant, Department of Radiology, Shifa International Hospital, Islamabad, Pakistan.
*Corresponding Author : Syed Murtaza H Kazmi
Chief of Department and Consultant, Department of Pulmonology, Shifa International Hospital, Islamabad, Pakistan.
Email: murtaza.kazmi@gmail.com
Received : Mar 29, 2025
Accepted : Apr 15, 2025
Published : Apr 22, 2025
Archived : www.jcimcr.org
Copyright : © Kazmi SMH (2025).
Abstract
Background: Chronic Hypersensitivity Pneumonitis (CHP) is a rare Interstitial Lung Disease (ILD) resulting from chronic immune-mediated inflammation and fibrosis due to recurrent exposure to inhaled antigens. Diagnosis becomes challenging when antigen exposure is unclear, resulting in delayed recognition, mismanagement, and potentially irreversible lung damage.
Case presentation: A 25-year-old man from a remote rural area of Pakistan presented with eight months of progressive breathlessness and non-productive cough. Initially misdiagnosed and mismanaged locally as idiopathic pulmonary fibrosis, he experienced progressive clinical deterioration, eventually becoming oxygen-dependent. Upon referral to our centre, a detailed clinical history revealed indirect avian exposure due to proximity to pigeons and hens, along with a concerning family history of respiratory illness—two siblings had died previously from similar conditions. High-Resolution Computed Tomography (HRCT) findings were atypical for classical CHP, prompting Video-Assisted Thoracoscopic (VATS)-guided lung biopsy. Histopathology demonstrated airway-centred fibrosis and bronchiolar metaplasia, confirming advanced-stage idiopathic CHP. Treatment with corticosteroids, mycophenolate mofetil, and pirfenidone was initiated, resulting in clinical stability at discharge and maintained improvement at a three-month follow-up. Discussion: This case illustrates the diagnostic challenges posed by CHP, particularly in the absence of a meaningful antigen exposure history and classic radiologic findings. Lung biopsy was instrumental in making the diagnosis. Moreover, the significant family history suggests possible (shared) genetic or environmental susceptibility, and genetic testing and family screening should be performed in these instances.
Conclusion: In the case of atypical ILD presentations, clinicians should maintain CHP as a potential diagnosis, the history should inquire for hereditary and indirect exposures, and multidisciplinary evaluations, including lung biopsy, should be pursued early in efforts to avoid misdiagnosis and inform effective management. This case emphasizes that genetic risk factors for CHP deserve studying.
Keywords: Chronic hypersensitivity pneumonitis; Interstitial lung disease; Familial ILD; Lung biopsy; Diagnostic uncertainty.
Citation: Kazmi SMH, Mirza W, Turab MS, Tariq AT, Dadan S, et al. Familial clustering in chronic hypersensitivity pneumonitis: Diagnostic dilemmas and role of lung biopsy. J Clin Images Med Case Rep. 2025; 6(4): 3564.
Introduction
Chronic Hypersensitivity Pneumonitis (CHP) is an Interstitial Lung Disease (ILD) resulting from chronic immune reactions to inhaled organic antigens, which cause chronic inflammation and fibrosis and, if untreated, respiratory failure [1,2]. However, a subset of patients does not have a well-defined, identifiable antigen exposure (which may delay diagnosis and management and result in irreversible pulmonary injury [3]. This clinical scenario emphasizes the importance of seeking indirect or less obvious exposures to environmental exposures. Diagnostic uncertainty is compounded by atypical radiological imaging presentation that warrants thorough evaluation, including histopathological confirmation with lung biopsy [4,5]. The genetic basis of CHP has been extensively studied in recent decades. Although the disease is mostly caused by environmental exposures, family clusters of ILD have been reported, suggesting an interaction between genetically-driven susceptibility and common environmental exposures [6,7]. It is difficult to differentiate genetic susceptibility and shared exposures in the household or the workplace when the history of exposure to the relevant antigen is unclear, thus highlighting the need for thorough family and genetic history assessments, especially in unexplained respiratory disease-prone populations [8,9]. Multidisciplinary assessments become critical in these complex scenarios, as misdiagnosis may result in prolonged morbidity and progression to advanced pulmonary fibrosis [10]. We report a case of a 25-year-old man from a remote rural area in Pakistan who developed progressive ILD initially mismanaged locally as idiopathic pulmonary fibrosis. On obtaining a detailed clinical history at our centre, indirect avian exposure and significant family history of unexplained respiratory illness led to suspicion of CHP. In light of the atypical CT imaging, the multidisciplinary team proposed a Video-Assisted Thoracoscopic Surgery (VATS)-guided lung biopsy that eventually confirmed idiopathic CHP. This report should serve to illuminate diagnostic challenges in atypical exposure and imaging findings cases, emphasise the need for multidisciplinary approach with biopsy, and underscore the necessity of further research analysing genetic and familial influences in CHP.
Case presentation
A 25-year-old man from a remote rural region of Balochistan came to our tertiary-care centre with a history of 8 months of worsening shortness of breath and dry cough. However, upon careful history-taking, it was found out that he had indirect exposure to birds as the patient lived near pigeons and hens but had no direct contact. Initially, he had a low-grade fever for about three days, and persistent breathlessness improved after five months with continuous oxygen supplementation. He had multiple admissions locally shortly before referral and had been poorly managed as progressive idiopathic pulmonary fibrosis with inappropriate choice and lack of antibiotics, with ongoing deterioration and ongoing oxygen dependency. On physical examination, he was thin, chronically ill, and required supplemental oxygen at 1.5 L/min. Lung auscultation was notable for bilateral, fine inspiratory crackles. He had no significant medical history apart from repeated hospital admissions for respiratory symptoms. Notably, his family history was concerning; two brothers previously died from similar unexplained respiratory illnesses at a young age. This unusual familial clustering raised suspicion of potential genetic susceptibility or shared environmental triggers, prompting consideration for genetic counseling and further familial screening. Laboratory investigations, including complete blood count, renal function tests, and autoimmune screenings, were unremarkable. Arterial blood gases confirmed hypoxemia (pH 7.39, PaCO₂ 50 mmHg, PaO₂ 60 mmHg) while on oxygen. High-Resolution Computed Tomography (HRCT) of the chest showed atypical features inconsistent with classical Chronic Hypersensitivity Pneumonitis (CHP). Findings included diffuse coarse interstitial thickening, traction bronchiectasis, and multiple small thin-walled cystic changes predominantly in upper lobes, resembling advanced fibrotic ILD with honeycombing (Figure 1). Due to unclear antigen exposure and atypical imaging findings, differential diagnoses included Idiopathic Pulmonary Fibrosis (IPF), connective tissue disease-associated ILD, and fibrotic Nonspecific Interstitial Pneumonia (NSIP). After detailed multidisciplinary discussions, Video-Assisted Thoracoscopic Surgery (VATS)-guided lung biopsy was performed to clarify the diagnosis. Histopathology revealed airway-centered fibrosis, bronchiolar metaplasia, chronic inflammatory infiltrates, architectural distortion, and fibroblastic plugs. No granulomas, fungal elements, or malignancy were found. These findings, combined with indirect exposure history, supported a diagnosis of idiopathic CHP complicated by advanced fibrosis. Initial hospital management included intravenous methylprednisolone pulse therapy, followed by oral prednisolone. Immunosuppressive therapy with mycophenolate mofetil and antifibrotic therapy with pirfenidone was started, along with co-trimoxazole prophylaxis against Pneumocystis Pneumonia (PCP). In the hospital, she had a pneumothorax requiring needle decompression and right-sided chest drain placement, with resolution without complication. Despite several attempts at oxygen weaning, the patient continued to have severe pulmonary fibrosis and persistent hypoxia. At discharge, the patient was stable but required ongoing supplemental oxygen. Discharge medications included oral prednisolone, mycophenolate mofetil, pirfenidone, co-trimoxazole, and calcium/vitamin D supplementation. The patient was counseled at length about avoiding the medication, compliance, follow-up, and reducing exposure to possible antigens. As future options, advanced therapeutic possibilities were contemplated, such as lung transplantation in case of the evolution of the disease. At the three-month follow-up, the patient showed clinical stability with no further deterioration. He remained oxygen-dependent at 1 liter per minute, tolerating medications without notable side effects. A repeat HRCT demonstrated stable fibrotic disease without further progression, suggesting a positive response to combined immunosuppressive and antifibrotic therapy. Further family assessment and genetic evaluation were advised, given the concerning familial history.
Histopathological analysis: Histopathological examination of the lung biopsy, stained with hematoxylin and eosin (H&E), demonstrated classic findings associated with Chronic Hypersensitivity Pneumonitis (CHP). Prominent bronchiolar metaplasia indicated extensive remodelling of airway epithelium, likely reflecting chronic antigen-driven epithelial injury. Peribronchiolar fibrosis, consistent with chronic inflammation and scarring, was frequently observed around the bronchioles. Significant architectural distortion with airway-centered interstitial fibrosis resulted in marked destruction of normal lung architecture. Patchy chronic inflammation characterized by lymphocytic infiltration was present, consistent with repeated immune-mediated injury triggered by antigen exposure. Fibroblastic plugs were also identified, indicative of abnormal tissue repair processes occurring within lung parenchyma. Notably, well-formed granulomas-typically seen in early-stage hypersensitivity pneumonitis-were absent, aligning with the chronic, fibrotic nature of the disease in this case. These histopathological findings, combined with radiological evidence of diffuse interstitial thickening and honeycombing, strongly supported advanced-stage CHP. The lung biopsy was instrumental in confirming the diagnosis, particularly given the significant overlap of clinical and radiological features with other fibrotic interstitial lung diseases (Figure 2).
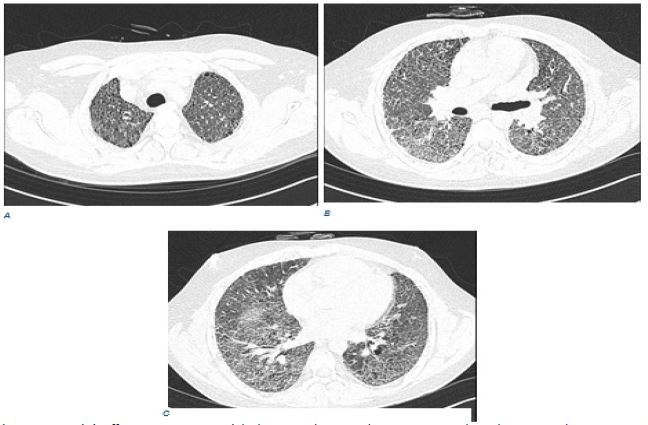
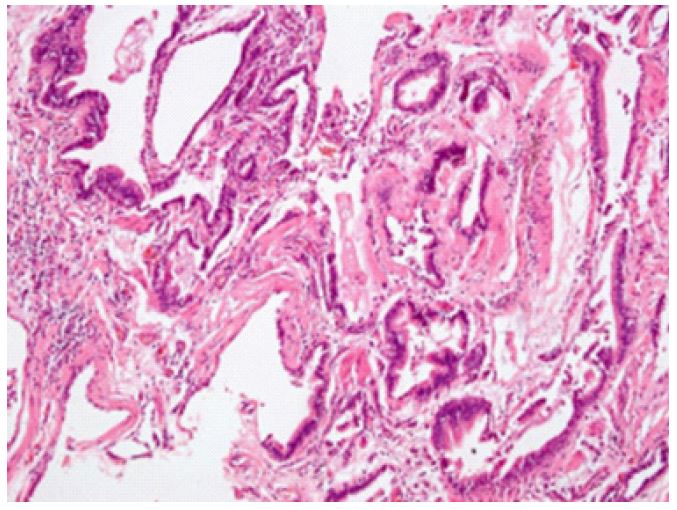
Table 1: Differentiating features between CHP, IPF, CTD-ILD, and NSIP.
| Feature | Chronic Hypersensitivity Pneumonitis (CHP) | Idiopathic Pulmonary Fibrosis (IPF) | Connective Tissue Disease-ILD (CTD-ILD) | Fibrotic Nonspecific Interstitial Pneumonia (NSIP) |
|---|---|---|---|---|
| Clinical Features | Dyspnea, cough, fatigue; history of antigen exposure | Progressive dyspnea, dry cough, clubbing | Dyspnea, cough, symptoms of underlying CTD (joint pain, Raynaud’s, etc) | Dyspnea, cough; generally milder than UIP |
| RadiologicalFeatures (HRCT) | Ground-glass opacities, centrilobular nodules, mid-upper lung predominance | Basal & subpleural honeycombing, reticulation (UIP pattern) | Variable (UIP, NSIP,OP patternsdepending on CTD) | Bilateral ground-glass opacities and reticulation |
| Histopathology | Lymphocytic inflammation, poorly formed granulomas, fibrosis | Usual Interstitial Pneumonia (UIP) with fibroblastic foci | Variable (UIP, NSIP, OP, DAD) depending on CTD | Homogeneous interstitial inflammation and fibrosis |
| Treatment | Antigen avoidance, steroids,immunosuppressants if needed | Antifibrotic (pirfenidone, nintedanib), lung transplant in severe cases | Immunosuppressants (steroids, DMARDs, biologics) | Immunosuppressants (steroids, azathioprine, mycophenolate) |
| Prognosis | Variable improves with antigen removal; fibrosiscan progress | Poor, median survival ~3–5 years | Depending on CTD severity; some respond wellto treatment | Better than UIP, but canprogress in some cases |
Discussion
Chronic Hypersensitivity Pneumonitis (CHP) typically results from chronic exposure to organic antigens, leading to inflammation, fibrosis, and progressive Interstitial Lung Disease (ILD) [1]. However, clinicians frequently encounter cases lacking classical exposure histories or typical radiological findings, as observed in this patient, posing significant diagnostic challenges. The absence of direct bird exposure and atypical HRCT imaging in our patient prompted further diagnostic evaluation, notably a lung biopsy, to establish a definitive diagnosis. In our case, radiological findings diverged from the classical ground-glass opacities and centrilobular nodules commonly seen in CHP, leading the multidisciplinary team toward a biopsy-based approach. Histopathology showed airway-centered fibrosis, bronchiolar metaplasia, chronic inflammatory infiltrates, and fibroblastic plugs. These findings supported a diagnosis of CHP despite the absence of granulomas, typically characteristic of early-stage hypersensitivity pneumonitis. Chronic variants of CHP often mimic other fibrotic ILDs, such as Idiopathic Pulmonary Fibrosis (IPF) or fibrotic nonspecific interstitial pneumonia (NSIP), complicating diagnosis. (Table 1) summarizes the key distinguishing features of CHP compared to other fibrotic interstitial lung diseases, emphasizing the critical role lung biopsy plays in complex diagnostic scenarios [4,5].
Familial clustering of ILD, as observed in our patient’s history, raises the possibility of genetic susceptibility or shared environmental exposure. Familial pulmonary fibrosis and telomeropathy syndromes, associated with mutations in Telomerase RNA Components (TERC), Telomerase Reverse Transcriptase gene (TERT), and poly(A)-specific ribonuclease (PARN), illustrate genetic predisposition in certain ILDs [6,7]. Although traditionally CHP is environmentally driven, growing evidence suggests genetic factors may influence disease susceptibility and progression, underscoring the importance of detailed family history-taking and genetic evaluation in patients with atypical presentations [8,9]. Given our patient’s strong familial history and clinical stability at three-month follow-up, further genetic investigations and family screening have been advised, which could enhance understanding of hereditary contributions in similar cases. Management of fibrotic CHP is challenging, particularly when progressive fibrosis leads to severe respiratory impairment. Current guidelines emphasize antigen avoidance as the primary management step, although complete avoidance can be challenging due to socioeconomic factors. While glucocorticoids may expedite initial recovery, their long-term benefit is uncertain. Alternative immunosuppressants such as azathioprine and mycophenolate mofetil may also be considered. In severe cases, antifibrotic therapy (e.g., pirfenidone or nintedanib) and lung transplantation have demonstrated favorable medium-term outcomes [10,11]. Our patient also required immunosuppressive therapy with a combination of corticosteroids and mycophenolate mofetil due to extensive fibrosis, as well as anti-fibrotic therapy with pirfenidone. The clinical stability of the patient, reflected by 3-month treatment stability on imaging, further reinforces the effectiveness of the therapeutic approach and highlights the importance of proactive, multidisciplinary management in achieving beneficial outcomes like those for our patient. Oxygen dependence despite optimized treatment reinforces the importance of close follow-up and timely consideration of advanced therapies such as lung transplantation. Early diagnosis and timely management greatly enhance prognosis, but the treatment for advanced fibrotic CHP is limited.
Conclusion
This case illustrates the diagnostic challenges of Chronic Hypersensitivity Pneumonitis (CHP) when classical exposure history and classical radiological features are missing. It emphasizes the necessity of multidisciplinary assessment along with lung biopsy to make an accurate diagnosis in complex presentations. It also brings to attention potential etiological factors such as genetic susceptibility and familial clustering as important aspects in the pathogenicity of CHP. Such initial manifestations together with short-term follow-up showing a favorable clinical response, strengthen the rationale for the use of combined immunosuppressive and antifibrotic treatment, highlighting the importance of early and appropriate therapy. Management is particularly complicated in patients with progressive fibrotic CHP, whose course requires long-term supplemental oxygen and possible advanced therapies such as lung transplantation. Clinicians should have a high suspicion for CHP in atypical ILD presentation, promote detailed environmental and family history, recommend genetic counselling and family screening when familial clustering is present and consider early histopathological confirmation in diagnostic uncertainties.
Declarations
Ethics approval and consent to participate: The Ethics Review Committee at Shifa International Hospital, Islamabad, Pakistan, approved this case report (IRB# 072-25). Written informed consent was obtained from the patient’s legal guardian for participation and publication.
Consent for publication: Written informed consent for publication of this case report, including clinical details and associated images, was obtained from the patient’s guardian. The consent document is available for editorial review upon request.
Competing interests: The authors declare no personal, financial, or other conflicts of interest related to this case report.
/Funding: No external funding was received for this work.
Authors’ contributions
Dr. Syed Murtaza H Kazmi: Conception of work, design of work; analysis and interpretation of data, drafting, reviewing it critically for important intellectual content
Wajahat Mirza: Conception of work, design of work; analysis and interpretation of data, drafting, reviewing it critically for important intellectual content
Dr. Ahmad Talha Tariq: Reviewing it critically for important intellectual content, final editing, and formatting.
Sajeel Turab: Reviewing it critically for important intellectual content, final editing, and formatting.
Dr. Sundus Dadan: Reviewing it critically for important intellectual content, final editing, and formatting.
Dr. Zafar Ali: Contribution to data collection, interpretation of clinical data, and assistance in drafting specific sections of the manuscript.
Dr. Rashed Nazir: Reviewing it critically for important intellectual content
Acknowledgments: We thank Shifa International Hospital, Islamabad, for supporting this work and enabling the multidisciplinary team management required for this complex case.
References
- Furusawa H, Cardwell JH, Okamoto T, Walts AD, Konigsberg IR, et al. Chronic hypersensitivity pneumonitis, an interstitial lung disease with distinct molecular signatures. Am J Respir Crit Care Med. 2020; 202(10): 1430-44.
- Rafique M, Arslan F, Khan J, Zaki S, Hussain A. Hypersensitivity Pneumonitis: An Interesting Case of Acute Shortness of Breath in a Young Patient. Cureus. 2024; 16(9): 68683.
- Rachid C, Hindi M, Fikri O, Amro L. Diagnostic Approach to Hypersensitivity Pneumonitis: A Report of Two Cases. Cureus. 2023; 15(8).
- Morell F, Villar A, Montero MÁ, Muñoz X, Colby TV, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013; 1(9): 685-94.
- Verma G, Jamieson F, Chedore P, Hwang D, Boerner S, et al. Hot tub lung mimicking classic acute and chronic hypersensitivity pneumonitis: two case reports. Can Respir J. 2007; 14(6): 354-6.
- Walters GI, Mokhlis JM, Moore VC, Robertson AS, Burge GA, et al. Characteristics of hypersensitivity pneumonitis diagnosed by interstitial and occupational lung disease multi-disciplinary team consensus. Respir Med. 2019; 155: 19-25.
- Mikumo H, Yanagihara T, Hamada N, Hashisako M, Ijichi K, et al. An autopsy case of bird-related chronic hypersensitivity pneumonitis presenting with repeated acute exacerbation. Respir Med Case Rep. 2018; 24: 92-4.
- Barnes H, Jones K, Blanc P. The hidden history of hypersensitivity pneumonitis. Eur Respir J. 2022; 59(1).
- Nogueira R, Melo N, e Bastos HN, Martins N, Delgado L, et al. Hypersensitivity pneumonitis: antigen diversity and disease implications. Pulmonology. 2019; 25(2): 97-108.
- Ionescu MD, Popescu NA, Stănescu D, Enculescu A, Bălgrădean M, et al. The Challenging Diagnosis of Interstitial Lung Disease in Children—One Case Report and Literature Review. J Clin Med. 2022; 11(22): 6736.
- Sorino C, Agati S, editors. Rare and Interstitial Lung Diseases-E-Book: Clinical Cases and Real-World Discussions. Elsevier Health Sciences. 2024.