
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Clinical Image - Open Access, Volume 6
Dyschromatosis universalis hereditaria with STRC- related sensorineural hearing loss in a child: A dual genetic diagnosis
Prasanth L1*; Alok Koshy Alexander1; Roohie Singh2; Angshuman Dutta3
1Resident, Department of ENT & HNS, Command Hospital (Eastern Command), Kolkata, India.
2Professor, Department of ENT & HNS, Command Hospital (Eastern Command), Kolkata, India.
3Professor, Department of ENT & HNS, Army Hospital (Research & Referral), New Delhi, India.
*Corresponding Author : Prasanth L
Resident, Department of ENT & HNS, Command Hospital (Eastern Command), Kolkata, India.
Tel: +91-9092660451;
Email: ragavprash28@gmail.com
Received : Apr 22, 2025
Accepted : May 09, 2025
Published : May 16, 2025
Archived : www.jcimcr.org
Copyright : © Prasanth L (2025).
Citation: Prasanth L, Alok Koshy A, Roohie S Angshuman D. Dyschromatosis universalis hereditaria with STRC- related sensorineural hearing loss in a child: A dual genetic diagnosis. J Clin Images Med Case Rep. 2025; 6(5): 3594.
Description
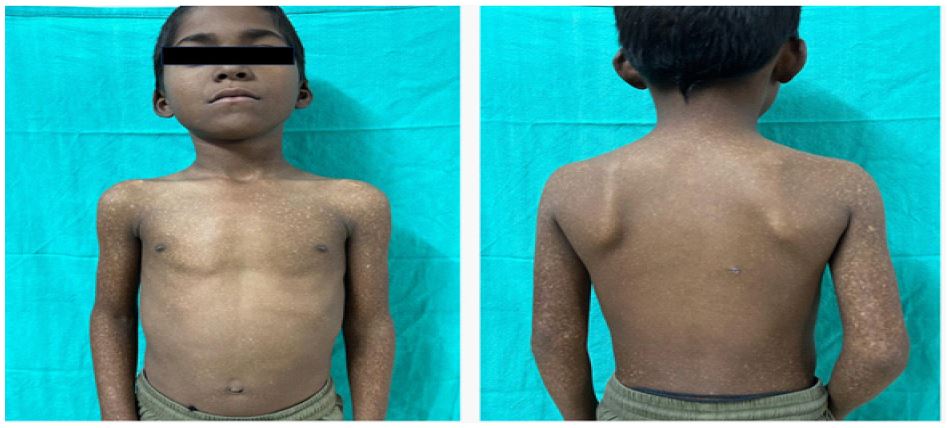
8-year-male child, born of a second- degree consanguineous marriage, a known case of moderate intellectual disability was referred from the paediatric outpatient department with complaints of bilateral hearing loss of insidious onset, gradually progressive initially noticed at the age of four years by his parents along with defective speech development. The hearing loss had become noticeable over time and is now significantly affecting his routine communication and academic activities. The child was born of a full-term pregnancy via caesarean section, with an unremarkable antenatal and perinatal history. There were no complications at birth and the neonatal period was uneventful. Developmentally, the child had delayed milestones and showed poor scholastic performance. Formal cognitive assessment revealed moderate intellectual disability associated with behavioural disturbances. There was no history of seizures or developmental regression. At around 01 year of age, parents noticed mottled skin pigmentation, which gradually spread to involve the face, trunk and extremities. The lesions were non-pruritic and consisted of interspersed hyperpigmented and hypopigmented macules with sparing of palms, soles and mucosa.
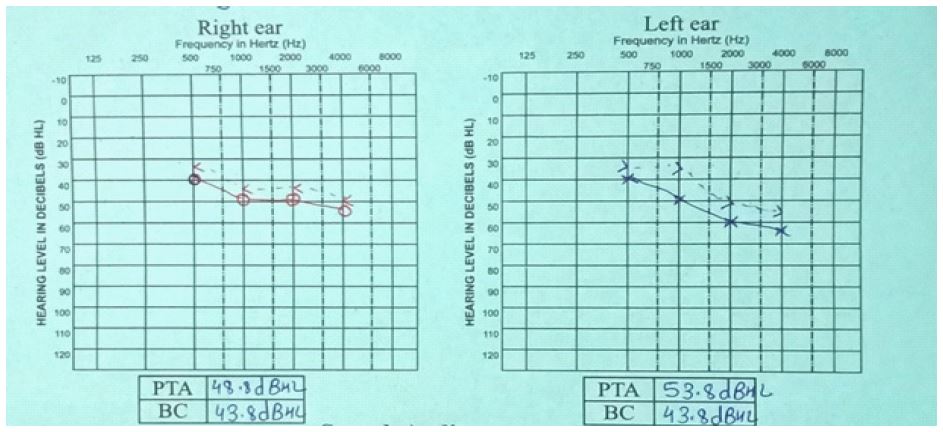
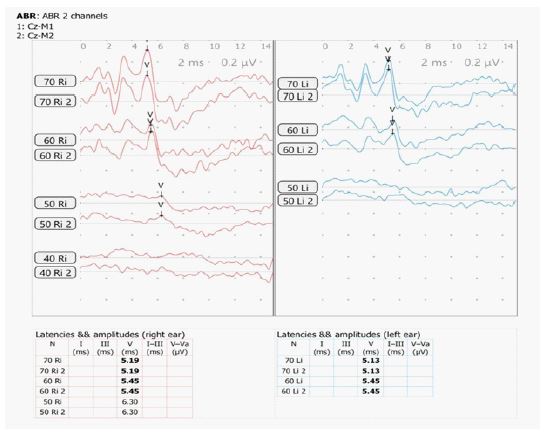
There is a negative family history of similar cutaneous, auditory or neurodevelopmental disorders. On examination, the child had diffuse, irregularly shaped, mottled pigmented macules over lower face, trunk and extremities (Figure 1). Systemic and neurological examinations were unremarkable. Otoscopic findings were normal. Pure tone audiometry (PTA) demonstrated bilateral moderate sensorineural hearing loss (Figure 2) and Brainstem Evoked Response Audiometry (BERA) showed wave V at 60 dBnHL on Right and at 50 dBnHL on left side (Figure 3). Punch biopsy from a hypopigmented lesion over back revealed basal layer pigment incontinence with dermal melanophages, consistent with Dyschromatosis Universalis Hereditaria (DUH). Genetic testing identified a homozygous a pathogenic mutation in the STRC gene (stereocilin), confirming a diagnosis of autosomal recessive non-syndromic sensorineural hearing loss (DFNB16 related). Though Genetic analysis for ABCB6 (ATP Binding Casette Subfamily B Member 6) was not done due to unavailability and high cost; nevertheless, the diagnosis of DUH was made clinically and histopathologically.
Discussion
DUH is a rare genodermatosis with autosomal dominant inheritance, rare reports have suggested potential systemic involvement, including neurological and auditory abnormalities. ABCB6 is the most commonly implicated gene in DUH and mutations in the transporter protein can also affect inner ear development, leading to hearing deficits [1]. STRC gene is associated with autosomal recessive non- syndromic hearing loss, specifically DFNB16 and its pathogenic variants are among the more common genetic causes of mild to moderate hearing loss [2]. Genetic studies are necessary to elucidate the various associations of DUH like sensorineural hearing loss [3].
This case illustrates a dual genetic diagnosis in a child involving clinically and histopathologically confirmed DUH and genetically confirmed STRC related non-syndromic hearing loss [4]. The co-existence of two genetically distinct conditions is likely attributable to consanguinity and highlights the importance of not assuming syndromic overlap when multiple symptoms are present. This case demonstrates the value of targeted genetic testing in establishing a precise diagnosis and guiding counselling.
Declarations
Conflicts of interest: Nil,
Informed consent: Informed consent was taken from the patient for this publication.
Funding: No funding was received to assist with the preparation of this manuscript. The authors have no relevant financial or non-financial interests to disclose.
References
- Al Hawsawi K, Al Aboud K, Ramesh V, Al Aboud D. Dyschromatopsia universalis hereditarian: Report of a case and review of the literature. Pediatr Dermatol. 2002; 19: 523-526.
- Baril SA, Wilson KA, Shaik MM, et al. The role of ATP-binding Cassette subfamily B member 6 in the inner ear. Nat Commun. 2024; 15: 9885.
- Redfield S, Shearer AE. STRC-Related Autosomal Recessive Hearing Loss. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. 2023; 1993-2025.
- Gupta, Mrinal MBBS, MD, DNB. Dyschromatosis universalis hereditaria with sensorineural hearing loss. Egyptian Journal of Dermatology and Venereology Jun. 2016;| 36(1): 26-27. DOI: 10.4103/1110-6530.194160.