
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 3
RLR-mediated IFN signaling and aberrant activation
Han Yang1,2; Yunran Feng1,2; Chunjing Feng2,3*; Xin Mu1,2
1 School of Pharmaceutical Science and Technology, Tianjin University, Tianjin 300072, China.
2 Tianjin University and Health-Biotech United Group, Joint Laboratory of Innovative Drug Development and Translational Medicine, Tianjin University, Tianjin 300072, China.
3 Health-Biotech Group Stem Cell Research Institute, Tianjin 301799, China.
*Corresponding Author: Chunjing Feng
Tianjin University and Health-Biotech United Group
Joint Laboratory of Innovative Drug Development
and Translational Medicine, Tianjin University, Tianjin 300072, China.
Email: fengchunjing@health-biotech.com
Received : Jan 16, 2022
Accepted : Feb 15, 2022
Published : Feb 22, 2022
Archived : www.jcimcr.org
Copyright : © Feng C (2022).
Abstract
Interferon (IFN) signaling is important for host cells in defending microorganisms. “Self” and “non-self” recognition is the key step for proper IFN functioning. Pathogen-derived double-stranded RNAs (dsRNAs) are immunogenic to the cytosolic sensor, RIG-I and MDA5. Together known as RIG-I-like receptors (RLRs), they form filament along the length of dsRNA substrate which in turn activate downstream cascades. Despite RLRs themselves and host encoded regulators managing to avoid self-intolerance, extensive studies in recent years uncovered that the boundary of “self” and “non-self” can be vague. Retrotransposon elements embedded in the genome of humans can turn into RLR ligands under certain circumstances, like incorrect RNA metabolism or epigenetic drug treatment, which in turn, lead to IFN signaling activation. The consequences of such activation can be different according to different circumstances. Here, we summarized the biological features of RLR-mediated IFN signaling and discussed aberrant IFN signaling through RLRs, including situations of gene mutations, irradiation, and anti-cancer drug treatment. We speculated that combining current cancer therapy and RLR-mediated IFN signaling activation would bring beneficial effects to cancer treatment.
Keywords: RLR; IFN; MDA5; RIG-I; ADAR1; DNA methylation.
Abbreviations: IFN: interferon; ISG, IFN-stimulated gene; PRR: pattern recognition receptor; PAMP: pathogen-associated molecular pattern; DAMP: damage-associated molecular pattern; cGAS: cyclic GMP-AMP synthase; dsRNA: double-stranded RNA; RIG-I: retinoic acid-inducible gene I; MDA5: melanoma differentiation-associated gene 5; TLR: tolllike receptor; ssRNA: single-stranded RNA; RLR: RIG-I-like receptor; GOF: gain-of-function; MAVS: mitochondrial antiviral-signaling protein; CARD: caspase activation and recruiting domain; CTD: C-terminal domain; lncRNA: long non-coding RNA; SeV: Sendai virus; VSV: vesicular stomatitis virus; EMCV: encephalomyocarditis virus; JEV: Japanese encephalitis virus; Poly I:C: polyinosinic–polycotylid acid; LMW: lower molecular weight; TRAF: tumor necrosis factor receptor-associated factor; TBK1: TANK Binding Kinase 1; IKK: inhibitor of NF-κB kinase; NEMO: NF-κB essential modulator; RNF135: Ring Finger Protein 135; TRIM65: tripartite motif containing 65; A: adenosine; I: inosine; LOF: loss-of-function; AGS: Aicardi–Goutières syndrome; WT: wildtype; SMS: Singleton-Merten syndrome; SLE: systemic lupus erythematosus; SPENCD: spondyloenchondromatosis; IR: ionizing radiation; 5-AZACdR: 5-aza-2’-deoxycytidine; ERV: endogenous retroviruses.
Citation: Yang H, Feng Y, Feng C, Mu X. RLR-mediated IFN signaling and aberrant activation. J Clin Images Med Case Rep. 2022; 3(2): 1682.
Introduction
Interferon (IFN) signaling pathway is an important defense mechanism for host cells to resist the invasion of microorganisms. It leads to the expression of IFN-stimulated genes (ISGs), including effectors that directly fight against pathogens and regulators modulating this stressed response as well as communicating with innate and adaptive immunity [1,2]. IFNs can be classified into three groups according to sequences, functions, and their receptors [3]. The type-I IFNs (IFN-I, e.g., IFN-a, IFN-b) and type-III IFNs (IFN-III, e.g., IFN-l) engage in anti-microorganism defense while type-II IFN (IFN-II, e.g., IFN-g) participates in regulating the host immunity [4,5].
All nucleated cells express receptors for IFN-I [6]. The proper functioning of this pathway is firstly dependent on the correct recognition of “self” and “non-self” molecules by the receptors. These proteins are called pattern recognition receptors (PRRs) as they recognize pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), instead of binding to specific sequences [7]. The “non-self” molecules can be DNA, RNA, lipoproteins, etc. Immunogenic nucleic acids can be derived exogenously from pathogenic infection or in vitro transcription/synthesis [8], or endogenously from incorrect DNA/RNA metabolisms [9]. Their corresponding PRRs are localized in the cytosol or on the lipid membranes of endosomes. The cytosolic sensors include DNA-recognizing cyclic GMP-AMP Synthase (cGAS) and double-stranded RNA (dsRNA)-responding Retinoic acid-inducible gene I (RIG-I) and Melanoma differentiation-associated gene 5 (MDA5) [10]. The endosomal toll-like receptors (TLRs) target DNA, single-stranded RNA (ssRNA) or dsRNA [7,11]. Because cellular DNAs are confined to the nucleus and mitochondria, pathogen-derived DNAs can be easily recognized as “non-self” in the endosome and cytoplasm [10]. Endogenous RNAs are mainly single-stranded, duplexes are not only short (e.g., IR-Alu is ~300 bp) but also have bulges and mismatches inside and are edited by ADAR1 to further impair the integrity [12]. In contrast, virus-derived dsRNAs are perfectly matched and can be much longer, making them an easy target for dsRNA sensors to bind. Endosomes are not supposed to have any DNA or RNA; thus, all forms of nucleic acids are targeted as “non-self” there. Of the receptors, RIG-I and MDA5 are together called RIG-I-like receptors (RLRs), sensing a range of RNA viruses infection[13], and their gain-offunction (GOF) mutations lead to autoimmune diseases due to the aberrant activation of type-I IFN signaling in the absence of pathogen invasion [14,15]. Here, we summarized RLR-mediated IFN signaling and current progress on the understanding of its aberrant activation.
RLR-mediated IFN signaling
Features of dsRNA binding
RIG-I and MDA5 share sequence and domain architecture similarities and use the same adaptor Mitochondrial antiviralsignaling protein (MAVS) for activating downstream cascades [10,16]. The N-terminus of RLR is a tandem caspase activation and recruiting domain (2CARDs) motifs mediating the signaling transduction; the central DExD/H box helicase domain in the middle and the C-terminal domain (CTD) are together responsible for dsRNA binding [17,18] (Figure 1A). Structural studies on RIG-I demonstrated that in the absence of dsRNA ligand, it exerts a conformation of auto-repressed manner, in which case the 2CARDs folds back to associate with helicase domain so that 2CARDs are not exposed to elicit signaling [19] (Figure 1B). A recent study suggested that endogenously encoded long noncoding RNA (lncRNA), lnc-Lsm3b, may contribute to the silence of RIG-I by sequestering it in a homeostasis state as well [20].
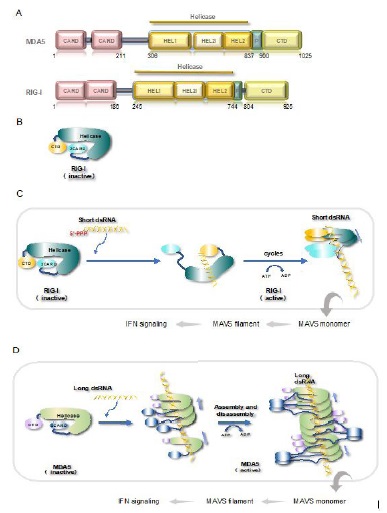
Although both receptors recognize and bind to dsRNAs, the binding patterns and preferred substrates are different. RIG-I recognizes the end of dsRNA bearing the 5’-ppp or 5’-pp structures through CTD then binds to it [21,22]. Next, this RIGI translocates to the interior of dsRNA upon hydrolyzing ATP through its helicase domain. The unoccupied 5’-end on dsRNA can recruit another molecule of RIG-I [23]. Cycles of ATP hydrolysis and translocation produce a RIG-I filament along the length of dsRNA [23] (Figure 1C). Such mode of action confers RIG-I is best stimulated by dsRNAs at a short length, e.g., 40-150 bp [24,25]. MDA5, on the other hand, binds to the interior of dsRNA as the speed-limiting step. New MDA5s bind quickly to this nucleation site and a filament is growing towards both sides [26]. Biochemical assays showed that MDA5 hydrolyses ATP for disassembly, but only the ones at the ends of filament are able to leave [26,27] (Figure 1D). This assembly-and-disassembly kinetics of binding determines that MDA5 prefers longer duplex RNAs, e.g., ~0.5-7 kb long dsRNA [25,26]. For both receptors, the ATPase activity was shown to serve as a proof-reading mean for correct substrates [26,28,29].
The differences in preference suggest that they are non-redundant dsRNA sensors. RIG-I is stimulated by negative-sense RNA viruses, like Sendai virus (SeV) and Vesicular Stomatitis Virus (VSV) [13,30], while MDA5 is intolerant to positive-sense RNA virus infection, such as picornavirus EMCV (Encephalomyocarditis virus) [13]. Besides, mice assays clearly showed that RIG-I is the responsible receptor for Japanese encephalitis virus (JEV) despite it being a positive-sense RNA virus [13]. RNA substrate determination has always been a challenging project for dsRNA-binding proteins. In the case of MDA5, it also binds to ssRNA randomly and with equivalent affinity in biochemical systems [31], which in turn, interferes with the analysis of assay results. One solution is to utilize the formation of filament, which encloses dsRNA substrate and thus protects it from RNase digestion in mild digesting conditions [12]. Yet, given that RLRs bind to dsRNA in a sequence-independent manner, bound RNAs in different viruses may have little sequence commonalities in general and thus is less informative. As noted, synthetic dsRNA polyinosinic–polycotylid acid (poly I:C) is composed of two nucleotides only, where the lower molecular weight (LMW) version with a shorter length is better in activating RIG-I and the longer version is best in stimulating MDA5 [25].
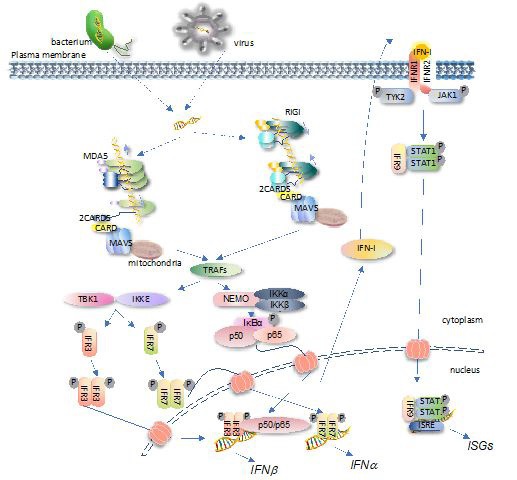
Downstream cascades
Albeit the filaments are formed by different mechanisms, they similarly induce the intermolecular 2CARDs forming oligomers [17,32]. The 2CARDs oligomers, in turn, recruit the mitochondria-anchoring protein MAVS for binding through the 2CARDsRLR-CARDMAVS interaction. Multiple MAVS are induced to form a prion-like aggregate via the MAVS-MAVS interaction through CARDMAVS motifs [17,33]. Such aggregation creates a new platform to recruit downstream factors, such as tumor necrosis factor receptor-associated factors (TRAFs) and the subsequently associated TANK Binding Kinase 1 (TBK1)/inhibitor of NF-κB kinase e (IKKe), NF-kB essential modulator (NEMO)/IKKa/ IKKb, etc. TBK1/IKKe and NEMO/IKKa/IKKb then activate the type-I IFN and NF-kB signaling respectively [34,35]. A schematic view of RLRs-mediated IFN signaling is illustrated in Figure 2.
A feature of RLR-mediated IFN signaling is the formation of filament, which arises from monomeric protein to large complex. Recent studies uncovered a post-filament event by hostencoded E3 ubiquitin ligases to regulate both RIG-I and MDA5 activity. RIPLET (also known as RNF135, Ring Finger Protein 135) and Tripartite Motif Containing 65 (TRIM65) were identified as RIG-I filament- and MDA5 filament-binding proteins, respectively [24,36,37]. Intriguingly, neither of the E3 ligases could bind to the corresponding RLR at monomeric form. Both ligases dictate the K63-linked ubiquitination of target RLRs, and such modification is required for RLR-mediated IFN activation.
Homeostasis and aberrant activation
dsRNA editing by ADAR1: As mentioned earlier, PAMPs or DAMPs are not sequence-dependent but pattern-dependent, which means that the silencing of IFN-I signaling in the absence of infection is very largely dependent on the control and homeostasis of cellular endogenous molecules [38]. For RLRs, such control comes from the modulation of the endogenous dsRNA pool. The adenosine deaminase acting on RNA 1 (ADAR1) is a dsRNA-specific adenosine deaminase that catalyzes adenosine (A) converting into inosine (I) in RNA duplex, such reaction melts the integrity of dsRNA structures by introducing mismatches (A: U becomes I_U) [39,40]. To be precise, ADAR1 controls the homeostasis of MDA5 [12,41]. Studies showed that the loss-ofexpression of ADAR1 results in the embryonic lethal phenotype in mice and is completely rescued by the additional loss-of-expression of MDA5 [41]. ADAR1 in humans is suggested to mainly edit endogenous inverted-repeat Alu (IR-Alu) elements [42,43], implicating that IR-Alu duplex is targeted by human MDA5 if without A-to-I editing. In line with this, loss-of-function (LOF) mutations of ADAR1 and GOF mutations of IFIH1 (encoding MDA5) identified in Aicardi–Goutières syndrome (AGS) patients were shown to aberrantly activate IFN signaling via the same endogenous dsRNA species, IR-Alu elements [12]. That is being said, the GOF mutants of MDA5 form signaling-transducing filament on IR-Alu duplexes even these RNAs are edited by ADAR1, or the wildtype (WT) MDA5 binds to unedited IR-Alu duplexes to activate IFN-I signaling, in the absence of viral infection.
AGS and SMS: AGS is an immune disorder affecting mainly skin and brain and is the prototype of type-I interferonopathies conceptualized firstly by Yanick J. Crow in 2011 [44]. The type-I interferonopathies are featured by the aberrant activation of IFN-I signaling in the absence of pathogen invasion, due to monogenic mutations of somatic genes and such mutations obey Mendel’s law [9,44]. Another example of such a disease is Singleton-Merten syndrome (SMS). Both RIG-I and MDA5 mutations were identified in SMS [14,15]. It is interesting that up to now, RIG-I mutations were not yet identified in AGS patients. Manifestations of SMS include but are not limited to, aortic calcification, dental dysplasia, and abnormalities in the skin [45,46]. Given that the GOF mutations of both receptors aberrantly activate IFN-I signaling to rely on the same downstream cascades, AGS and SMS share some manifestations clinically, for example, glaucoma [47]. In fact, as the aberrant activation of IFN-I signaling is a feature in type-I interferonopathies, their symptoms were shown to have some overlapped manifestations. Phenotypic overlaps were also seen in AGS and systemic lupus erythematosus (SLE) [48], AGS and spondyloenchondromatosis (SPENCD) [49], etc. An excellent review on this topic can be found in the following reference [50]. We summarized RLR gene mutations in Table 1 [15,51-62].
Table 1:RLR mutations identified in AGS and/or SMS. Gene and mutation sites are described, as well as the belonging domains.
Gene |
Mutation |
Domain |
Reference |
DDX58 |
c.803G>T (p. Cys268Phe) |
Helicase |
51 |
c.1118A>C (p. Glu373Ala) |
Helicase |
51 |
|
c.1529A>T (p. Glu510Val) |
Helicase |
52 |
|
c.1551G>C (p. Gln517His) |
Helicase |
53 |
|
IFIH1 |
c.992C>G (p. Thr331Arg) |
Helicase |
54 |
c.992C>T (p. Thr331Ile) |
Helicase |
54 |
|
c.1009A>G (p. Arg337Gly) |
Helicase |
55 |
|
c.1114C>T (p. Leu372Phe) |
Helicase |
56 |
|
c.1165G>A (p. Gly389Arg) |
Helicase |
14 |
|
c.1178A>T (p.Asp393Val) |
Helicase |
55 |
|
c.1178A>C (p. Asp393Ala) |
Helicase |
14 |
|
c.1331A>G (p. Glu444Gly) |
Helicase |
14 |
|
c.1347C>G (p. Asn449Lys) |
Helicase |
14 |
|
c.1354G>A (p. Ala452Thr) |
Helicase |
56 |
|
c.1465G>A (p. Ala489Thr) |
Helicase |
57 |
|
c.1465G>T (p. Ala489Ser) |
Helicase |
58 |
|
c.1483G>A (p. Gly495Arg) |
Helicase |
55 |
|
c.1747A>G (p. Ile583Val) |
Helicase |
14 |
|
c.2156C>T (p. Ala719Val) |
Helicase |
14 |
|
c.2159G>A (p. Arg720Gln) |
Helicase |
55 |
|
c.2335C>T (p. Arg779Cyc) |
Helicase |
55 |
|
c.2336G>A (p. Arg779His) |
Helicase |
55 |
|
c.2336G>T (p. Arg779Leu) |
Helicase |
14 |
|
c.2390 A>T (p. Asp797Val) |
Helicase |
59 |
|
c.2342G>A (p. Gly781Glu) |
Helicase |
14 |
|
c.2407A>T (p. Ile803Phe) |
Helicase |
14 |
|
c.2439A>T (p. Glu813Asp) |
Helicase |
60 |
|
c.2465G>A (p. Arg822Gln) |
Helicase |
61 |
|
c.2471G>A (p. Arg824Lys) |
Helicase |
14 |
|
c.2486C>G (p. Thr829Ser) |
Helicase |
14 |
|
c.2544T>G (p. Asp848Glu) |
Helicase |
62 |
|
c.2561T>A (p. Met854Lys) |
Helicase |
14 |
|
c.2866A>G (p. Ile956Val) |
CTD |
14 |
|
c.2936T>G (p. Leu979Trp) |
CTD |
14 |
From the table, it is shown that mutations in RIG-I and MDA5 occurred in motifs responsible for dsRNA-recognition and binding, the helicase domain and CTD, implicating these GOF mutants break the immune balance by losing tolerance to the endogenous self RNAs. The primate-specific retrotransposon, IR-Alu, was identified in human cells as the ligand for GOF MDA5 in 2018 [12], yet the endogenous ligands for GOF RIG-I are not identified so far. It is noted that the embryonic lethal effect in mice by deficiency of ADAR1 was rescued by MDA5 or MAVS knockout, but not RIG-I [41,63,64], implicating that the dsRNAs edited by ADAR1 are not targeted by RIG-I. Moreover, take IR-Alu as an example, it normally locates inside the intron in nucleus or 3’-UTR of mRNAs [12,42] where no 5’-ppp or 5’- pp end is exposed for RIG-I binding. Thus, the endogenous RNA substrates for GOF MDA5 and RIG-I are different. Evidence suggested that under certain circumstances such as ionizing radiation (IR) therapy, small nuclear RNAs U1 and U2 can translocate to the cytoplasm and be targeted by RIG-I to activate IFN signaling [65]. This observation suggested that the nuclear noncoding RNAs may have the ability to activate RIG-I, however, under normal circumstances, they are confined to the nucleus. Whether other non-coding RNAs present in cytoplasm become the ligand of GOF RIG-I is of interest to know.
Impacts of DNA methylation: Homeostasis breakdown not only happens in the case of gene mutations but also occurs in the treatment of pathological conditions, especially cancer chemotherapies and irradiation (as mentioned previously). DNA methylation plays important role in regulating DNA transcription epigenetically. Methylation and transcription are generally negatively correlated with each other in the case of transposon elements [66,67]. An epigenetic anti-cancer drug, 5-aza-2’-deoxycytidine (5-AZA-CdR), inhibits DNA methylation and is used in the treatment of hematological malignancies [68]. Evidence demonstrated that MDA5 is the key player in the effectiveness of 5-AZA-CdR in killing cancer cells [69,70]. Furthermore, the interplay between ADAR1 and MDA5 determines the efficacy of anti-cancer drug treatment, where ADAR1 acts as negative feedback for IFN activation [71]. All these observations implicated that endogenous transposon elements can induce IFN-I signaling when activated in transcription. In line with this, inhibition of LSD1 activity in breast tumors was reported to induce IFN-I signaling which in turn, beneficial to antitumor treatment [72]. LSD1 stands for lysine-specific demethylase 1 and is a histone H3K4 demethylase. Trimethylated H3K4 histone is found in actively transcribed regions [73] where LSD1 suppresses gene expression by converting trimethylated H3K4 to mono- or unmethylated one [74]. Data showed that both sense-strand and antisense-strand endogenous retroviruses (ERV) (belong to retrotransposon) transcription were increased in LSD1-KO cells, thus producing endogenous dsRNA species that activate MDA5 in the cytoplasm [75].
In a word, we now are fully aware that the concept “self” and “non-self” are not based on the origins of molecules. Changed conditions such as chemotherapy or gene mutations can transfer the “self” genome into a reservoir of stimulant ligands, and to lead undesired consequences to the cell and the host.
Outlooks
A combining effort of biochemistry, biophysics, cellular approaches, and in vivo assays for almost two decades has unmasked the biology of the two cytosolic dsRNA sensors. Given that RLR-mediated IFN signaling is a stress response, it is regulated at multiple steps by a variety of host-encoded regulators. The recent findings that two E3 ligases (RIPLET and TRIM65) specifically bind to the filamentous form of the corresponding RLRs but not the monomeric form to regulate substrate protein’s activity unleashed an open question of whether such mode of action represents a new kind of mechanism by which the cells apply to regulate PRRs. Biochemical data showed that at least several members of the TRIM family are capable of binding to different RNA helicases in a conformation-selecting manner (LGP2-TRIM14, Dicer-TRIM25, etc.) [37].
Mounting evidence from studies on autoimmune diseases and anti-cancer therapies demonstrated that our genome not only contains genes necessary for cellular survival and proliferation but also is embedded with repeated elements that are potentially immunogenic when transcribed in certain conditions. These repeated elements are retrotransposons integrated into the human genome as a remanence from an ancient age. Evolutions have shaped RLRs with balanced capacities between sensitivity and selectivity. Under this logic, as others had suggested, the IFN pathway is more like a pathway hosts utilize to monitor cellular status, rather than inhibiting pathogen invasion only. PAMPs and DAMPs then may also be divided into the following two groups: first, wrong molecules (such as perfect, unedited dsRNA) derived endogenously or exogenously; second, right molecules in the wrong place (such as DNA in cytoplasm or ssRNA in endosome).
In the case of AGS raised from mutations of MDA5, treatment, for now, focuses on restricting the IFN signaling pathway, for example, the use of JAK inhibitors (ruxolitinib, baricitinib, etc.) [9,76,77], or antibodies against IFN-a [78]. Blocking the recognition of the IR-Alu duplex by GOF MDA5 is definitely a very important direction to investigate, but it is technically hard. RNAs are normally knocked down by treatment of siRNA or miRNA [79]. However, this is difficult for IR-Alu RNAs because these elements are pre-formed duplexes. Furthermore, the enormous number of Alu elements in the genome [80,81] makes it impossible to knockout by the CRISPR-Cas9 system. Yet it is worth noting that IR-Alu duplexes are not perfectly matched. The edition by ADAR1 further melts the integrity of these duplexes [12,82]. Thus, bulges and mismatches are present in every single IR-Alu and can be potential sites for unwinding.
For the repeated elements in the genome, on the one hand, they have the potential to elicit unwanted stress signaling; on the other hand, they can be manipulated to serve as good tools in killing cancer cells. Indeed, in the study of LSD1, it was suggested that deficiency of LSD1 overcomes tumor resistance to PD-1 antibody treatment in mice [75]. Thus, the combining of anti-tumor antibodies, immune cells, and epigenetic drugs would become a promising direction of cancer therapy in the future.
Declarations
Funding: This work was supported by the State Key Laboratory of Veterinary Etiological Biology, CAAS, and the Guangdong Provincial Key Laboratory of Precision Medicine and Clinical Translation Research of Hakka Population (Grants SKLVEB2020KFKT001 and 2018B030322003KF03 to X.M.)
Conflicts of interest: The authors declare no conflicts of interest.
Author contributions: H.Y., Y.F., C.F., and X.M. wrote and revised the manuscript, H.Y. draw the figures.
References
- WM Schneider. Chevillotte CM. Rice, Interferon-Stimulated Genes: A Complex Web of Host Defenses, Annu. Rev. Immunol. 2014; 32: 513-545.
- S Hervas-Stubbs, JL Perez-Gracia, A Rouzaut, MF Sanmamed, A Le Bon, et al. Direct Effects of Type I Interferons on Cells of the Immune System, Clin Cancer Res. 2011; 17: 2619-2627.
- H Zhou, S Chen, M Wang, A Cheng. Interferons and Their Receptors in Birds: A Comparison of Gene Structure, Phylogenetic Analysis, and Cross Modulation, IJMS. 2014; 15: 21045-21068.
- S Li, M Gong, F Zhao, J Shao, Y Xie, et al. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection, Cell Physiol Biochem. 2018; 51: 2377-2396.
- Z Dembic. Cytokines in Innate Immunity, in: Reference Module in Biomedical Sciences, Elsevier. 2021: B9780128187319001000.
- G Schreiber. The molecular basis for differential type I interferon signaling, Journal of Biological Chemistry. 2017; 292: 7285-7294.
- JCH Tam, DA Jacques. Intracellular immunity: finding the enemy within--how cells recognize and respond to intracellular pathogens, Journal of Leukocyte Biology. 2014; 96: 233-244.
- X Mu, S Hur. Immunogenicity of In Vitro -Transcribed RNA, Acc. Chem. Res. 2021; 54: 4012-4023.
- MA Lee-Kirsch. The Type I Interferonopathies, Annu. Rev. Med. 2017; 68: 297-315.
- A Ablasser, S Hur. Regulation of cGAS- and RLR-mediated immunity to nucleic acids, Nat Immunol. 2020; 21: 17-29
- K Takeda, S Akira. Toll‐Like Receptors, Current Protocols in Immunology. 2015; 109: 109.
- S Ahmad, X Mu, F Yang, E Greenwald, JW Park, et al. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation, Cell. 2018; 172: 797-810.e13.
- H Kato, O Takeuchi, S Sato, M Yoneyama, M Yamamoto, et al, Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses, Nature. 2006; 441: 101-105.
- C Lu, M MacDougall. RIG-I-Like Receptor Signaling in SingletonMerten Syndrome, Front. Genet. 2017; 8: 118.
- GI Rice, S Park, F Gavazzi, LA Adang, LA Ayuk, et al. Genetic and phenotypic spectrum associated with IFIH1 gain‐of‐function, Human Mutation. 2020; 41: 837-849.
- Q Sun, L Sun, HH Liu, X Chen, RB Seth, et al. The Specific and Essential Role of MAVS in Antiviral Innate Immune Responses, Immunity. 2006; 24: 633-642.
- B Wu, A Peisley, D Tetrault, Z Li, EH Egelman, et al. Molecular Imprinting as a Signal-Activation Mechanism of the Viral RNA Sensor RIG-I, Molecular Cell. 2014; 55: 511-523.
- S Hur. Double-Stranded RNA Sensors and Modulators in Innate Immunity, Annu. Rev. Immunol. 2019; 37: 349-375.
- E Kowalinski, T Lunardi, AA McCarthy, J Louber, J Brunel, et al. Structural Basis for the Activation of Innate Immune PatternRecognition Receptor RIG-I by Viral RNA, Cell. 2011; 147: 423- 435.
- M Jiang, S Zhang, Z Yang, H Lin, J Zhu, et al. Self-Recognition of an Inducible Host lncRNA by RIG-I Feedback Restricts Innate Immune Response, Cell. 2018; 173: 906-919.e13.
- M Schlee, A Roth, V Hornung, CA Hagmann, V Wimmenauer, et al. Recognition of 5′ Triphosphate by RIG-I Helicase Requires Short Blunt Double-Stranded RNA as Contained in Panhandle of Negative-Strand Virus, Immunity. 2009; 31: 25-34.
- V Hornung, J Ellegast, S Kim, K Brzózka, A Jung, et al. 5’-Triphosphate RNA Is the Ligand for RIG-I, Science. 2006: 314: 994-997.
- A Peisley, B Wu, H Yao, T Walz, S Hur. RIG-I Forms Signaling-Competent Filaments in an ATP-Dependent, Ubiquitin-Independent Manner, Molecular Cell. 2013: 51: 573-583.
- C Cadena, S Ahmad, A Xavier, J Willemsen, S Park, et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity, Cell. 2019; 177: 1187-1200.e16.
- H Kato, O Takeuchi, E Mikamo-Satoh, R Hirai, T Kawai, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene 5, Journal of Experimental Medicine. 2008; 205: 1601-1610.
- A Peisley, MH Jo, C Lin, B Wu, M Orme-Johnson, et al. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments, Proceedings of the National Academy of Sciences. 2012; 109: E3340-E3349.
- Y del Toro Duany, B Wu, S Hur. MDA5-filament, dynamics and disease, Current Opinion in Virology. 2015; 12: 20-25.
- SC Devarkar, B Schweibenz, C Wang, J Marcotrigiano, SS Patel. RIG-I Uses an ATPase-Powered Translocation-Throttling Mechanism for Kinetic Proofreading of RNAs and Oligomerization, Molecular Cell. 2018; 72: 355-368.e4.
- Q Yu, K Qu, Y Modis. Cryo-EM Structures of MDA5-dsRNA Filaments at Different Stages of ATP Hydrolysis, Molecular Cell. 2018; 72: 999-1012.e6.
- J Rehwinkel, CP Tan, D Goubau, O Schulz, A Pichlmair, et al. RIGI Detects Viral Genomic RNA during Negative-Strand RNA virus Infection, Cell. 2010; 140: 397-408.
- A Peisley, C Lin, B Wu, M Orme-Johnson, M Liu, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition, Proceedings of the National Academy of Sciences. 2011; 108: 21010-21015.
- B Hur. How RIG-I like receptors activate MAVS, Current Opinion in Virology. 2015; 12: 91-98.
- F Hou, L Sun, H Zheng, B Skaug, QX Jiang, et al MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response, Cell. 2011; 146: 448-461.
- J Sohn, S Hur. Filament assemblies in foreign nucleic acid sensors, Current Opinion in Structural Biology. 2016; 37: 134-144.
- V Hornung. Snap Shot: Nucleic Acid Immune Sensors, Part 1, Immunity. 2014; 41: 868-868.e1.
- X Lang, T Tang, T Jin, C Ding, R Zhou, et al TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity, Journal of Experimental Medicine. 2017; 214: 459-473.
- K Kato, S Ahmad, Z Zhu, JM Young, X Mu, et al. Structural analysis of RIG-I-like receptors reveals ancient rules of engagement between diverse RNA helicases and TRIM ubiquitin ligases, Molecular Cell. 2021; 81: 599-613.e8.
- D Tang, R Kang, CB Coyne, HJ Zeh, MT Lotze. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity, Immunol Rev. 2012; 249: 158-175.
- A Bhate, T Sun, JB Li. ADAR1: A New Target for Immuno-oncology Therapy, Molecular Cell. 2019; 73: 866-868.
- Q Wang, X Li, R Qi, T Billiar. RNA Editing, ADAR1, and the Innate Immune Response, Genes. 2017; 8: 41.
- BJ Liddicoat, R Piskol, AM Chalk, G Ramaswami, M Higuchi, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself, Science. 2015; 349: 1115-1120.
- H Chung, JJA Calis, X Wu, T Sun, Y Yu, et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown, Cell. 2018; 172: 811-824.e14.
- G Ramaswami, R Zhang, R Piskol, LP Keegan, P Deng, et al. Identifying RNA editing sites using RNA sequencing data alone, Nat Methods. 2013; 10: 128-132.
- YJ Crow. Type I Interferonopathies: A novel set of inborn errors of immunity: Type I interferonopathies, Annals of the New York Academy of Sciences. 2011; 1238: 91-98.
- BB Gay, JP Kuhn. A Syndrome of Widened Medullary Cavities of Bone, Aortic Calcification, Abnormal Dentition, and Muscular Weakness (The Singleton—Merten Syndrome), Radiology. 1976; 118: 389-395.
- A Feigenbaum, C Müller, C Yale, J Kleinheinz, P Jezewski, et al. Singleton-Merten syndrome: An autosomal dominant disorder with variable expression, Am. J. Med. Genet. 2013; 161: 360- 370.
- YJ Crow, DS Chase, J Lowenstein Schmidt, M Szynkiewicz, GMA Forte, et al. Rice, Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR and IFIH1, Am. J. Med. Genet. 2015; 167: 296-312.
- YJ Crow. Cree encephalitis is allelic with Aicardi-Goutières syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism, Journal of Medical Genetics. 2003; 40: 183-187.
- TA Briggs, GI Rice, S Daly, J Urquhart, H Gornall, et al. Tartrateresistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature, Nat Genet. 2011; 43: 127-131.
- YJ Crow, N Manel. Aicardi-Goutières syndrome and the type I Interferonopathies, Nat Rev Immunol. 2015; 15: 429-440.
- MA Jang, EK Kim, H Now, NTH Nguyen, WJ Kim, et al. Mutations in DDX58, which Encodes RIG-I, Cause Atypical SingletonMerten Syndrome, The American Journal of Human Genetics. 2015; 96: 266-274.
- L Prasov, BL Bohnsack, AS El Husny, LC Tsoi, B Guan, et al. DDX58 (RIG-I)-related disease is associated with tissue-specific interferon pathway activation, J Med Genet. 2021.
- CR Ferreira, YJ Crow, WA Gahl, PJ Gardner, R Goldbach-Mansky, et al. DDX58 and Classic Singleton-Merten Syndrome, J Clin Immunol. 2019; 39: 75-80.
- LM de Carvalho, G Ngoumou, JW Park, N Ehmke, N Deigendesch, et al. Musculoskeletal Disease in MDA5-Related Type I Interferonopathies: A Mendelian Mimic of Jaccoud’s Arthropathy: Musculoskeletal Disease In Mda-5-Related Type I Interferonopathies, Arthritis & Rheumatology. 2017; 69: 2081-2091.
- GI Rice, Y Del Toro Duany, EM Jenkinson, GM Forte, BH Anderson, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling, Nat Genet. 2014; 46: 503-509.
- H Oda, K Nakagawa, J Abe, T Awaya, M Funabiki, et al. AicardiGoutières Syndrome Is Caused by IFIH1 Mutations, the American Journal of Human Genetics. 2014; 95: 121-125.
- AC Bursztejn, TA Briggs, Y del Toro Duany, BH Anderson, JO Sullivan, et al. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: Overlap between Aicardi-Goutières and Singleton-Merten syndromes, Br J Dermatol. 2015; 173: 1505-1513.
- W Xiao, J Feng, H Long, B Xiao, ZH Luo. Case Report: AicardiGoutières Syndrome and Singleton-Merten Syndrome Caused by a Gain-of-Function Mutation in IFIH1, Front Genet. 2021; 12: 660953.
- K Hasegawa, H Tanaka, N Futagawa, H Miyahara, Y Higuchi, et al. A novel pathogenic variant p. ASP797VAL in IFIH1 in a Japanese boy with overlapping SINGLETON‐MERTEN syndrome and AICARDI‐GOUTIÈRES syndrome, Am J Med Genet. ajmg.a. 2021: 62478.
- S Amari, K Tsukamoto, A Ishiguro, K Yanagi, T Kaname, et al. An extremely severe case of Aicardi-Goutières syndrome 7 with a novel variant in IFIH1, Eur J Med Genet. 2020; 63: 103646.
- M Pettersson, B Bergendal, J Norderyd, D Nilsson, BM Anderlid, et al. Further evidence for specific IFIH1 mutation as a cause of Singleton-Merten syndrome with phenotypic heterogeneity, Am. J. Med. Genet. 2017; 173: 1396-1399.
- L Ruaud, GI Rice, C Cabrol, J Piard, M Rodero, et al. Autosomaldominant early-onset spastic paraparesis with brain calcification due to IFIH1 gain-of-function, Human Mutation. 2018; 39: 1076- 1080.
- NM Mannion, SM Greenwood, R Young, S Cox, J Brindle, et al. The RNA-Editing Enzyme ADAR1 Controls Innate Immune Responses to RNA, Cell Reports. 2014; 9: 1482-1494.
- K Pestal, CC Funk, JM Snyder, ND Price, PM Treuting, et al. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development, Immunity. 2015; 43: 933-944.
- DRE Ranoa, AD Parekh, SP Pitroda, X Huang, T Darga, et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs, Oncotarget. 2016; 7: 26496-26515.
- WA Schulz, C Steinhoff, AR Florl. Methylation of Endogenous Human Retro elements in Health and Disease, in: W Doerfler, P Böhm (Eds.), DNA Methylation: Development, Genetic Disease and Cancer, Springer Berlin Heidelberg. 2006: 211-250.
- CP Walsh, JR Chaillet, TH Bestor. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation, Nat Genet. 1998; 20: 116-117.
- HC Tsai, H Li, L Van Neste, Y Cai, C Robert, et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells, Cancer Cell. 2012; 21: 430-446.
- D Roulois, H Loo Yau, R Singhania, Y Wang, A Danesh, et al. DNADemethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts, Cell. 2015; 162: 961-973.
- KB Chiappinelli, PL Strissel, A Desrichard, H Li, C Henke, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses, Cell. 2015; 162: 974-986.
- P Mehdipour, SA Marhon, I Ettayebi, A Chakravarthy, A Hosseini, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency, Nature. 2020; 588: 169-173.
- Y Qin, SN Vasilatos, L Chen, H Wu, Z Cao, et al. Huang, Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade, Oncogene. 2019; 38: 390-405.
- H Santos-Rosa, R Schneider, AJ Bannister, J Sherriff, BE Bernstein, et al. Active genes are tri-methylated at K4 of histone H3, Nature. 2002; 419: 407-411.
- Y Shi, F Lan, C Matson, P Mulligan, JR Whetstine, et al. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1, Cell. 2004; 119: 941-953.
- W Sheng, MW LaFleur, TH Nguyen, S Chen, A Chakravarthy, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade, Cell. 2018; 174: 549-563.e19.
- N König, C Fiehn, C Wolf, M Schuster, E Cura Costa, et al. Familial chilblain lupus due to a gain-of-function mutation in STING, Ann Rheum Dis. 2017; 76: 468-472.
- ML Frémond, MP Rodero, N Jeremiah, A Belot, E Jeziorski, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173 -activating mutations in 3 children, Journal of Allergy and Clinical Immunology. 2016; 138: 1752-1755.
- F De Ceuninck, F Duguet, A Aussy, L Laigle, P Moingeon. IFN-α: A key therapeutic target for multiple autoimmune rheumatic diseases, Drug Discovery Today. 2021; 26: 2465-2473.
- RW Carthew, EJ Sontheimer. Origins and Mechanisms of miRNAs and siRNAs, Cell. 2009; 136: 642-655.
- Initial sequencing and analysis of the human genome, Nature. International Human Genome Sequencing Consortium. 2001; 409: 860-921.
- MA Batzer, PL Deininger. Alu repeats and human genomic diversity, Nat Rev Genet. 2002; 3: 370-379.
- LE Rieder, RA Reenan. The intricate relationship between RNA structure, editing, and splicing, Seminars in Cell & Developmental Biology. 2012; 23: 281-288.