
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Acute brain injury after pulmonary nodule surgery with ornithine transcarboxylase deficiency: A case report and literature review
Jiamei Liu1; Zhiqiong Zuo1; Shaojun Hu2; Xiaoling Huang1*
1Department of Comprehensive Surgical Ward, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
2Department of Pathology, Zhejiang Provincial Hospital of Chinese Medicine, Hangzhou, China.
*Corresponding Author : Xiaoling Huang
Department of Comprehensive Surgical Ward, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Email: huangxiaoring@163.com
Received : Apr 03, 2023
Accepted : Apr 27, 2023
Published : May 04, 2023
Archived : www.jcimcr.org
Copyright : © Huang X (2023).
Abstract
Ornithine Trans Carboxylase Deficiency (OTCD) is a rare congenital genetic related disease.
Domestic and foreign researches on OTCD are relatively rare till now. The present study reports a case of OTCD deficiency with acute brain injury after pulmonary nodule surgery, and analyzes its pathogenesis, clinical manifestations, diagnosis and treatment based on the literatures, with the aim to provide guidance for clinical practice.
Keywords: Ornithine trans carboxylase deficiency; Urea cycle disorder; Acute brain injury; Hyperammonemic ncephalopathy.
Citation: Liu J, Zuo Z, Hu S, Huang X. Acute brain injury after pulmonary nodule surgery with ornithine transcarboxylase deficiency: A case report and literature review. J Clin Images Med Case Rep. 2023; 4(5): 2399.
Introduction
Ornithine Trans Carboxylase Deficiency (OTCD) is a rare congenital genetic metabolic disease. Patients usually present with interrupted urea cycle, increased blood ammonia, decreased blood citrulline and arginine, increased glutamate and alanine, while increased urinary orotic acid as well pyrimidine excretion, which requiring special nursing care [1,2]. Among them, late-onset OTCD mainly occurs after the neonatal period. Although the incidence is low, due to the lack of specific clinical features, OTCD suffers are often misdiagnosed as neonatal sepsis, neonatal hypoxic-ischemic encephalopathy, birth trauma, food poisoning, gastric Intestinal disease, epilepsy, hepatic encephalopathy, intracranial infection, Reye syndrome, neurodegenerative disease or schizophrenia, etc [3-5]. OTCD with severe complications can even be rapidly life-threatening, though previous studies have shown that the onset of OTCD was extremely rare in adulthood [6]. Here we report the clinical characteristics of a critically ill patient with acute brain injury after pulmonary nodule surgery caused by delayed-onset OTCD and review the literatures to provide guidance for clinical diagnosis and treatment.
Case presentation
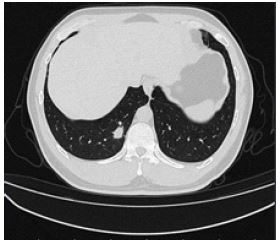
A 42-year-old male was admitted to the General Thoracic Surgery Department of our hospital on April 19, 2022 due to “one month’s lung space-occupying lung”. Past history: a history of hypertension for more than 3 years, oral administration of amlodipine besylate tablets 1# QD, blood pressure can be controlled, and occasional dizziness and discomfort in the past. One month ago, the lung shadow was found in the physical examination of the local hospital, and anti-inflammatory treatment was given for 10 days. Allergy history: penicillin skin test positive; cefminox skin test negative; VB1 skin test positive; Supp deep skin test positive. On April 19, our hospital reexamined chest CT: nodules in the basal segment of the right lower lung (Figure 1). The doctor recommended surgical treatment, and the patient’s family agreed to the surgical treatment.
On the afternoon of April 20, a wedge resection of the right lower lobe was performed, and the patient’s preoperative mental state was normal. Postoperative pathological findings: chronic granulomatous inflammation. At 17:30 in the after noon, the patient returned to the ward after the operation. The postoperative analgesic pump was hydromorphone/NS 6 mg/100 ml, and the continuous dosage was 1 ml/h. Subsequently, parecoxib 40 mg was given intravenously, and the following drugs were administered intravenously: cefminox 2G, azasetron 10 mg, aminocaproate sodium chloride 4G, aminocaproate sodium chloride 4G, rabeprazole 20 mg , Compound three-dimensional B needle 2 (B1, B6, B12). At around 21:30 in night, the patient became emotionally agitated.
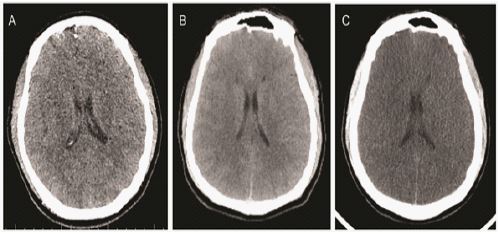
Early morning on April 21, the patient’s symptoms of emotional agitation were not relieved. Considering delirium, 10 mg of diazepam was administered intramuscularly, and the symptoms were relieved. Three hours later, with persistent delirium and agitation, the patient was given haloperidol 5 mg + scopolamine 0.15 mg IM. At around 8:00 in the morning, the department of Mental Health, Anesthesiology, and Infectious Diseases consulted the patient, and the conclusion was: postoperative delirium, but cerebrovascular accident and organic disease cannot be ruled out, and head CT or head MR examination was recommended. Medication adjustment: moxifloxacin was discontinued, quetiapine 50 mg -100 mg/once per night, haloperidol 5 mg + scopolamine 0.3 mg/intramuscular injection. Cranial CT examination results: There were no definite abnormal signs on the brain CT plain scan, and organic diseases were excluded (Figure 2A).
On April 22, the patient’s condition began to deteriorate. In the morning, the patient’s mood was still stable, the consciousness was clear. The call could be simply answered correctly, and the occasionally drowsiness. However, starting in the afternoon, the patient became confused, had paroxysmal restlessness, disordered speech, and sometimes answered inappropriately, without obvious impulsive behavior. On the morning of April 23, the patient’s condition further deteriorated. Physical examination: blood pressure 182/91 mmHg (1 mmHg=0.133 kPa), unconsciousness, lethargic state, pupils equal in size, presence of light, no response to stimuli of limbs, right Pap sign was not elicited, left Pap sign was positive. Then one-stop CTA+perfusion was performed to evaluate the cerebrovascular condition. Head CT report: no obvious signs of emergency (Figure 2B).
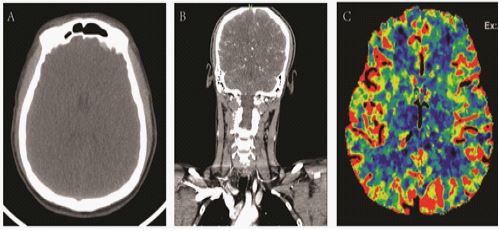
CT perfusion imaging of cranial blood vessels (NCCT): Brain parenchyma showed swelling (Figure 3A), and head MR+DWI examination were suggestion. CTA: Right vertebral artery slender. The A1 segment of the right anterior cerebral artery was thinned (Figure 3B). CTP: There was no significant decrease in bilateral parenchymal perfusion (Figure 3C). Nursing measures: changing the mask to inhale oxygen, using Ringer’s intravenous drip, rechecking blood gas after volume supplementation (blood gas can be sent to ICU), monitoring oxygen saturationand monitor blood sugar. Emergency plasma ammonia determination: blood ammonia 498.10 μmol/L. At about 8:00 PM, the suffer was transferred to the intensive care unit, and symptomatic treatment was received such as lowering intracranial pressure and blood ammonia, and performed CRRT at the bedside, after which the blood ammonia continued to decrease.
On April 25, the patient’s condition continued to deteriorate. Head CT display (Figure 2C): brain tissue has diffuse swelling, edema and reduced density. Pseudo subarachnoid hemorrhage is waiting to be discharged. Compared with the previous film (2022-4-23, Figure 2B), the lesions have obviously progressed. Subsequently, the department of neurology, neurosurgery, infection, anesthesia, radiology, digestive, pathology, kidney disease center, and severe medicine conducted emergency consultations, and gave positive opinions as well treatment and nursing measures. Department of Infectious
Diseases: 1. Eliminate the low primary urea synthase; 2. Lumina 0.06 TID decreased indirect bilirubin experimentally; 3. Check the possibility of poison.
Anesthesiology: 1. Consider metabolic diseases, congenital enzyme function is low, especially urea synthase; 2. Eliminate the possibility of poisons; 3. Stop taking exogenous amino acids. It is suggested that efforts should be made to maintain neural function; Strength the treatment of reducing intracranial pressure; Strengthen the treatment of reducing blood ammonia.
Gastroenterology: The blood ammonia is increased, and the primary liver damage is excluded. The low urea synthase and other diseases are considered.
Radiology: Brain tissue of head CT has diffuse swelling, edema and reduced density, which is obviously progressive (Figure 2C); CT reexamination is recommended after treatment.
Pathology: The pathological lung tissue after operation showed chronic granulomatous inflammation, which was considered to be fungal infection, and cerebrospinal fluid examination was still needed to exclude cryptococcal infection. Severe medicine department: Ammonia metabolism disorder, viral encephalitis, cryptococcal encephalitis and poisoning are not excluded. It is suggested to add fluconazole, midazolam, propofol, muscle relaxants for maintenance, stop amino acid and poison identification, and transfer to the comprehensive care unit.
Neurosurgery: Diffuse brain swelling, poor prognosis, no indication for surgical treatment. It is recommended that the internal medicine department continue conservative treatment, strengthen dehydration, reduce brain edema, and actively search for the cause.
Neurology: Considering the possibility of metabolic encephalopathy, heredity or metabolic disease in combination with significant abnormal blood ammonia, it is recommended to improve the relevant enzyme or gene examination. At present, dehydration and antiepileptic treatment are used to avoid the use of drugs that affect blood ammonia (KaipuLan, sodium valproate, carbamazepine).
Kidney disease center: At present, the patient has high blood ammonia and hypernatremia. It is recommended to continue renal replacement therapy. Be careful to use other nephrotoxic drugs during the course of the disease, and pay attention to monitoring renal function and electrolytes. At 16:00 on April 25, the patient was transferred to the comprehensive ICU for further active treatment and nursing. The genome of cerebrospinal fluid pathogens was sequenced and OTC genetic genes were screened.
Gene detection and screening results: The Metagenomics sequencing (DNA+RNA) of the CSF pathogen in this patient was negative. The sequencing and screening of the whole exon of the patient’s genetic gene showed that there was a pathogenic missense mutation in OTC gene (NM-000531.6) (Figure 4). The mutation of G into A at 622 nucleotide position of chromosome (X: 38262952) leads to the change of alanine encoded at this position into threonine, NM_ 000531.6(OTC):c. 622G>A (p.Ala208Thr). It was reported, this missense mutation mainly leaded to ornithine transcarboxylase deficiency (OTCD) [7,8]. The heterotopic site as a pathogenic variation has been included ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/variation/ 97274/).According to the gene detection results, combined with the previous clinical manifestations and diagnosis, we finally determined that the patient was an OCTD suffer.
Discussion
Pathogenesis
Ornithine transcarbamylase (OTC, MIM # 311250, RefSeq NP_000522) is a mitochondrial urea cycle enzyme that can cleave the reaction between carbamoyl phosphate and ornithine to form citrulline and phosphate [9]. OTC deficiency is the most common urea cycle enzyme defect, belonging to X-linked inheritance. OTC is the second enzyme in the urea cycle. Ornithine and carbamoyl phosphate generate citrulline under the action of OTC. In this reaction process, the lack of OTC will cause congenital metabolic diseases [6]. OTC deficiency inhibits the metabolism of carbamoyl phosphate, resulting in a large increase of whey acid in urine. Uridine and uracil can also be detected in urine. OTCD is a congenital metabolic disease caused by inhibition of carbamoyl phosphate metabolism. The patient’s urea circulation was interrupted, blood ammonia increased, blood citrulline and arginine decreased, glutamine and alanine increased, while urinary whey acid and pyrimidine excretion increased, and finally encephalopathy occurred [10]. Research shows that a large number of ammonia molecules produced by amino acid degradation quickly form glutamine with glutamic acid in brain cells and accumulate in brain cells, which increases its osmotic pressure and leads to brain cell edema. Brain edema not only causes insufficient blood supply, but also damages the function of neurons, axons, dendrites and synapses, leading to a series of brain metabolism and neurochemical abnormalities, resulting in the corresponding clinical symptoms: hyperammonemic encephalopathy [11]. OTCD is a rare congenital genetic metabolic disease, with a prevalence rate of 1/14000 – 1/77000, especially in adults [9]. Michael et al. searched PubMed, MEDLINE and Google Scholar databases and found that from 1987 to 2016, there were only 30 adult OTCD cases reported [6]. It is worth noting that although the disease is extremely rare in adults, its mortality rate is as high as 30% [6].
Clinical manifestation
The most common clinical manifestations of OTCD are mental state changes, seizures, brain edema and death [9]. The manifestations of various enzyme deficiency in the urea cycle are mainly nervous system symptoms caused by hyperammonemia, but the symptoms vary greatly among different types or among different patients of the same type. The closer the enzyme defect is to the beginning of the urea cycle, the more severe the symptoms are. Hyperammoniaemia is the main manifestation among various urea cycle enzyme deficiency diseases. Hyperammoniaemia can lead to a variety of brain dysfunction, including encephalopathy, epilepsy, brain edema, intracranial hypertension, coma and death [6]. The onset age of the disease can be from neonatal to adult [12]. The serum urea nitrogen level was usually lower than l mg/dl, and the blood ammonia concentration was higher; CT brain scanning can find brain edema, but most of the children were misdiagnosed as lung disease, sepsis or intracranial hemorrhage, which led to improper treatment and premature death. In our above cases, the patient’s mental state changed, and postoperative delirium occurred. The blood ammonia concentration was up to 498.10 μ Mol/L, CT also showed brain edema; all of the characteristics were relatively consistent. Studies have shown that late onset OTCD patients can be seen in all age stages, and the incidence in infancy may be related to the change from breast-feeding to regular milk (containing high protein) feeding. Elder children or adults may be caused by eating high protein. In late onset OTCD manifestations, patients may have encephalopathy attacks after emergencies, such as diet changes, acute diseases, trauma, pregnancy and use of corticosteroids or valproic acid [6,13,14]. In this case, the predisposing factors of the patientmay be high protein diet and diet change. The patient likes to keep fit at ordinary times, and his diet is often high in protein. His diet structure changes suddenly after surgery. Research shows that the incidence of OTCD in the United States and Finland is 1:17 000 and 1:60 000 respectively [15,16], of which the neonatal incidence is 1:8000 [12]. Male patients showed severe symptoms. Most of the newborn male OTCD patients could not survive, while female patients were heterozygous, showing different symptoms. However, female patients with severe OTCD may have Hyper Ammonemia (HA) or nervous system symptoms, which are easy to be misdiagnosed and cause high mortality. Therefore, pregnant women carrying OTCD gene should carry out gene testing of placental villi and amniotic fluid cells to determine the fetal status and decide whether to continue pregnancy [17].
Diagnosis and genetic variation
According to the literature, any patient with unexplained neurological and mental diseases accompanied by selective anorexia, as well as coma without obvious causes of brain edema and respiratory alkalosis, should consider the diagnosis of this disease [18]. OTCD diagnosis is mainly based on clinical symptoms, blood ammonia level and other general biochemical tests, such as blood amino acids, urine organic acids, gene detection [3]. For patients with mental state changes, seizures, brain edema, etc., the first thing is to measure the concentration of ammonia in the blood. Except for tissue hypoxia, liver and kidney function maintain normal function, but there are exceptions [5]. Secondly, the concentration of amino acids in blood was measured. The values of ornithine, glutamine and alanine in OTCD patients increased, while the values of citrulline and arginine decreased. The concentrations of amino acids and organic acids in the urine of patients can also be used as reference values. In addition to high blood ammonia, the concentration of serum saratin in patients’ urine can also be used as a clinical auxiliary diagnosis [5,19,20].
Elevated blood ammonia is the main abnormal indicator of OTCD patients, and OTC gene variation is another key diagnostic factor of OTCD. So far, more than 530 OTC gene mutations have been reported, but no hot spot mutations have been found [3,5]. OTC deficiency is the most common genetic defect in the urea cycle. OTCD is an X-linked recessive genetic disease. As a result, almost all hemizygous males will suffer from this disease. OTC gene is expressed in liver and small intestine, but complete urea cycle expression and activity only occur in liver. The human OTC gene is located on the X chromosome in the Xp21.1 band. Ten exons and nine introns span 73 kb, forming the human OTC gene with an open reading frame of 1062 nucleotides. More than 400 pathogenic mutations have been identified [10]. The diagnosis of OTC deficiency based on DNA detection is through the sequencing of OTC exons and intron/exon boundaries. OTC deficiency in clinically suspected patients is as high as 80% - 90% [6,10,21]. In diagnosis, the increase of urinary chylous acid occurs simultaneously with the increase of serum ammonia and alanine, with a sensitivity of 91% and a specificity of 70% [6,22]. Therefore, OTC gene mutation detection can more accurately diagnose OTCD. The cases reported in this article were initially misdiagnosed as other diseases, missed the opportunity of diagnosis and treatment, and finally developed into acute brain injury due to the failure to measure the concentration of blood ammonia and various amino acids in time and to sequence OTC genes as early as possible after the early onset of symptoms.
Therapeutic method
The main treatment for acute hyperammonemia caused by OTC deficiency is to limit the supplement of protein, arginine or citrulline in diet; High calorie support to promote anabolic state (insulin supplement as required); and promote alternative ways to transfer amino acid nitrogen from the urea cycle through nitrogen scavenging drugs [23-25]. The initial treatment for hyperammonemia is to stop all protein intake for at least 24 to 48 hours, restart intake at 0.25 to 0.5 g/kg, and then titrate. If the OTCD patient has a liver coma with hyperammonemia ≥ 300 μ mol/L, hemodialysis and treatment for alternative nitrogen metabolism pathway and arginine or/and citrulline should be used immediately to control the blood ammonia level to avoid damage to the brain [23,26,27].
Sodium benzoate and sodium phenylbutyrate transfer amino acid nitrogen from urea cycle by alternative way. About 40% benzoate combines with glycine and converts to hippuric acid, and 50% to 90% phenylbutyrate combines with glutamine to form phenylacetylglutamine, which is then excreted from the urine. It was reported that intravenous injection of sodium benzoate and sodium phenylacetatehad been proved to improve the survival rate of patients with hyperammonemia crisis. Patients weighing more than 25 kg should receive sodium phenylacetate and sodium benzoate (5500 mg each) as load dose within 90 to 120 minutes. The maintenance dose was given within 24 hours. The infusion should be diluted with 10% glucose before administration, with a dilution of 1:10. Arginine infusion shall not exceed 150 mg/kg/h [6]. However, regarding the long-term safety of sodium benzoate and its use in pregnant patients, its prescription in pregnant patients is still unclear. Therefore, special attention should be paid when taking sodium benzoate during pregnancy. Lamb et al. reported that a 29 year old pregnant woman gave birth successfully after taking sodium benzoate without any adverse reactions [28]. Similarly, Li and other researchers gave a 28 year old pregnant woman oral sodium benzoate without adverse reactions, and achieved the delivery of a healthy baby [17]. These researches indicate that sodium benzoate is effective and relatively safe for both mothers and infants in the treatment of OTCD during pregnancy. However, the safety of sodium benzoate during pregnancy needs to be further confirmed by large sample size and long-term follow-up studies.
In addition, patients with hyperammonemia may need to monitor epileptic seizure activity with EEG according to their clinical status. It is reported that the orthooxidation of valproic acid produces 2-propyl-4-valeric acid, which inhibits carbamoyl phosphate synthetase I, thereby inhibiting the enzymatic step before the urea cycle, leading to hyperammonemia. Therefore, valproic acid is contraindicated in OTC deficiency. Some cases show that the ammonia level increases significantly after using valproic acid [29].
Conclusion
OTCD is the main cause of congenital hyperammonemia. It can be asymptomatic before onset, often caused by infection, high protein diet and other factors. The clinical symptoms are lack of specificity, the disease progresses rapidly, and it can die from acute hyperammonemia encephalopathy, which is very easy to be misdiagnosed. Therefore, timely diagnosis and emergency treatment are very important to reduce the high mortality rate. Early diagnosis, individualized diet, drug treatment and liver transplantation are the main strategies to reduce the mortality and disability rate of OTCD patients. Rapid control of blood ammonia level is the key to reduce the mortality rate. For patients with unexplained vomiting, consciousness disorders, mental disorders, acute and chronic encephalopathy, blood ammonia should be detected in time, blood amino acids and urine organic acids should be analyzed, and OTC gene detection, early diagnosis and early treatment should be carried out as soon as possible. Family surveys and genetic counselling may be an indispensable aspect of prevention. The proband and parents should carry out OTC gene analysis of the family as soon as possible, so that the identification of the pathogenic gene is helpful for genetic counseling and early diagnosis.
Declarations
Ethical approval: Its study was conducted according to the Declaration of Helsinki, and approval was obtained from the Medical Ethics Committee at The First Affiliated Hospital of Zhejiang University (approval number: IIT20220753A).
Competing interests: The authors declare no competing interests.
Zuo and Hu collected data and prepared figures. All authors reviewed the manuscript.
Funding: Not applicable.
Availability of data and materials: Not applicable.
References
- Rüegger CM, Lindner M, Ballhausen D, Baumgartner MR, Beblo S, J. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders [J]. Journal of inherited metabolic disease. 2014; 37: 21-30.
- Batshaw ML, Tuchman M, Summar M, Seminara J. A longitudinal study of urea cycle disorders [J]. Molecular genetics and metabolism. 2014; 113: 127-130.
- Wang LP, Luo HZ, Song M, Yang ZZ, Yang F, et al. Hemizygous deletion in the OTC gene results in ornithine transcarbamylase deficiency: A case report [J]. World journal of clinical cases. 2022; 10: 1417-1422.
- Posset R, Garbade S F, Boy N, Burlina A B, Dionisi-Vici C, et al. Transatlantic combined and comparative data analysis of 1095 patients with urea cycle disorders-A successful strategy for clinical research of rare diseases [J]. 2019; 42: 93-106.
- Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision [J]. Journal of inherited metabolic disease. 2019; 42: 1192-1230.
- Pizzi MA, Alejos D, Hasan TF, Atwal PS, Krishnaiengar SR, et al. Adult Presentation of Ornithine Transcarbamylase Deficiency: 2 Illustrative Cases of Phenotypic Variability and Literature Review [J]. The Neurohospitalist. 2019; 9: 30-36.
- Sánchez AI, Rincón A, García M, Suárez-Obando F. Urea Cycle Defects: Early-Onset Disease Associated with A208T Mutation in OTC Gene-Expanding the Clinical Phenotype [J]. 2017; 2017.
- Daijo K, Kawaoka T, Nakahara T, Nagaoki Y, Tsuge M, et al. Late-onset ornithine transcarbamylase deficiency associated with hyperammonemia [J]. Clinical journal of gastroenterology. 2017; 10: 383-387.
- Lu D, Han F, Qiu W, Zhang H, Ye J, et al. Clinical and molecular characteristics of 69 Chinese patients with ornithine transcarbamylase deficiency [J]. Orphanet journal of rare diseases. 2020; 15: 340.
- Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update [J]. Journal of genetics and genomics = Yi chuan xue bao. 2015; 42: 181-194.
- Braissant O. Current concepts in the pathogenesis of urea cycle disorders [J]. Molecular genetics and metabolism, 2010; 100: S3-S12.
- Gordon N. Ornithine transcarbamylase deficiency: A urea cycle defect [J]. European journal of paediatric neurology : EJPN: official journal of the European Paediatric Neurology Society. 2003; 7: 115-121.
- Brassier A, Gobin S, Arnoux J B, Valayannopoulos V, Habarou F, et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: A series of 90 patients [J]. Orphanet journal of rare diseases. 2015; 10.
- Cordero D R, Baker J, Dorinzi D, Toffle R. Ornithine transcarbamylase deficiency in pregnancy [J]. Journal of inherited metabolic disease. 2005; 28: 237-240.
- Keskinen P, Siitonen A, Salo M. Hereditary urea cycle diseases in Finland [J]. Acta paediatrica (Oslo, Norway: 1992), 2008; 97: 1412-1419.
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy [J]. Advances in pediatrics, 1996; 43(127-170).
- Li l, Liu Y, Du J. Analysis of treatment regimen in a pregnant patient with ornithine transcarbamylase deficiency [J]. Journal of Chinese Pharmaceutical Sciences. 2020; 20: 666-670.
- Plöchl W, Plöchl E, Pokorny H, Kozek-Langenecker S, Zacherl J, et al. Multiorgan donation from a donor with unrecognized ornithine transcarbamylase deficiency [J]. Transplant international: Official journal of the European Society for Organ Transplantation. 2001; 14: 196-201.
- Ellaway CJ, Bennetts B, Tuck RR, Wilcken B. Clumsiness, confusion, coma, and valproate [J]. Lancet (London, England). 1999; 353: 1408.
- Nicolaides P, Liebsch D, Dale N, Leonard J, Surtees R. Neurological outcome of patients with ornithine carbamoyltransferase deficiency [J]. Archives of disease in childhood. 2002; 86: 54-56.
- Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human Ornithine Transcarbamylase (OTC) gene [J]. Human mutation. 2006; 27: 626-632.
- Grünewald S, Fairbanks L, Genet S, Cranston T, Hüsing J, Leonard JV, Champion MP, et al. How reliable is the allopurinol load in detecting carriers for ornithine transcarbamylase deficiency? [J]. Journal of inherited metabolic disease. 2004; 27: 179-186.
- Cederbaum S, Lemons C, Batshaw ML. Alternative pathway or diversion therapy for urea cycle disorders now and in the future [J]. Molecular genetics and metabolism. 2010; 100: 219-220.
- Batshaw ML, MacArthur RB, Tuchman M. Alternative pathway therapy for urea cycle disorders: Twenty years later [J]. The Journal of pediatrics. 2001; 138: S46-54.
- Kido J, Kawasaki T, Mitsubuchi H, Kamohara H, Ohba T, et al. Hyperammonemia crisis following parturition in a female patient with ornithine transcarbamylase deficiency [J]. World journal of hepatology, 2017; 9: 343-348.
- Schaefer F, Straube E, Oh J, Mehls O, Mayatepek E. Dialysis in neonates with inborn errors of metabolism [J]. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 1999; 14: 910-918.
- Feillet F, Leonard JV. Alternative pathway therapy for urea cycle disorders [J]. Journal of inherited metabolic disease. 1998; 21: 101-111.
- Lamb S, Aye C Y, Murphy E, Mackillop L. Multidisciplinary management of Ornithine Transcarbamylase (OTC) deficiency in pregnancy: Essential to prevent hyperammonemic complications [J]. BMJ case reports. 2013; 2013.
- Lheureux PE, Penaloza A, Zahir S, Gris M. Science review: Carnitine in the treatment of valproic acid-induced toxicity - what is the evidence? [J]. Critical care (London, England). 2005; 9: 431-440.