
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Giant-cell glioblastoma in a child: A rare case report and literature review
Nadia Bouzid; Amal Chamsi*; Syrine Lahiouel; Sarra Sghaier; Sameh Tebra
Medical School Sousse, Radiotherapy Department, University of Sousse, Farhat Hached Hospital, Sousse, Tunisia.
*Corresponding Author : Amal Chamsi
Medical School Sousse, Radiotherapy Department, University of Sousse, Farhat Hached Hospital, Sousse, Tunisia.
Email: chamsiamal90@gmail.com
Received : Apr 10, 2023
Accepted : May 05, 2023
Published : May 12, 2023
Archived : www.jcimcr.org
Copyright : © Chamsi A (2023).
Abstract
In Pediatric Giant Cell Glioblastoma (GCG) as a sub type of Glioblastoma (GBM), is a rare variant. However, it has a better prognosis.
We report à case of a 12-year-old male child treated for GCG in the radiation oncology department, Farhat Hached hospital, Sousse, Tunisia in 2021.
The child presented with a progressively worsening headache and vomiting. On neurological examination, he had left kinetic cerebellar ataxia. A magnetic resonance imaging displayed a left cerebellar hemisphere mass. The patient had a tumor total resection. The diagnosis of GCG was confirmed by histological study. The postoperative course was uneventful and the CT brain scan was without residual tumor. The child underwent Radiotherapy (RT) at a dose of 59.4 Gy with concurrent daily and adjuvant Temozolomide chemotherapy. Five months after the treatment, he presented with symptomatic intradural metastasis, spinal cord compression and leptomeningeal involvement. He received palliative RT at a dose of 8 Gy and he remained stable.
Supposedly to the literaure review, our patient’s history is marked by an unusual evolution with unfavorable prognosis. Future studies are still needed to establish a consensual management of this entity.
Keywords: Giant-cell glioblastoma; Irradiation; Intradural metastasis; Prognosis.
Citation: Bouzid N, Chamsi A, Lahiouel S, Sghaier S, Tebra S. Giant-cell glioblastoma in a child: A rare case report and literature review. J Clin Images Med Case Rep. 2023; 4(5): 2412.
Introduction
Pediatric Brain Tumors (BT) are the most common solid tumors in children and the second among all childhood malignancies after leukemia [1].
Glioblastoma Multiforme (GBM) or grade IV gliomas-tumors is the most frequent primary BT [2]. However, the incidence in children is very low, 0.8 per 100,000 children [3]. It is a clinically, histologically and genetically quite heterogeneous, highly malignant tumor [4].
Giant Cell Glioblastoma (GCG) as a sub type of GBM [5], is a rare variant with an incidence of 0.8% of BT and 5% of GBM. Supposedly, it has a better prognosis.
We report a case of GCG in a 12-year-old boy along with a clinicopathological and therapeutic description and literature review.
Case presentation
A 12-year-old male child presented with a two-month history of progressively worsening headache and vomiting. On neurological examination, the child was conscious, had left kinetic cerebellar ataxia and multiple caféaulait spots in the trunk. A contrast-enhanced CT scan showed an intra axial mass in the left cerebellar region with significant surrounding edema causing mass effect and onset of amygdala engagement.
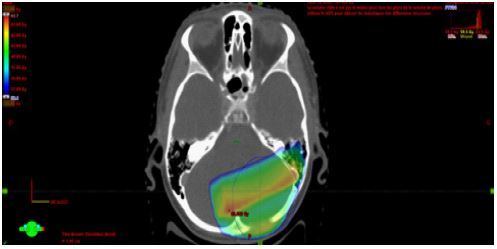
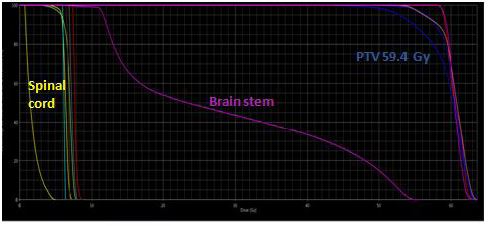
To further evaluate the lesion, a Magnetic Resonance Imaging (MRI) was performed and displayed a left cerebellar hemisphere mass, 25 x 20 mm in diameter with cystic component, lacking perilesional vasogenic edema. The mass was heterogenous, hypointense on T1-weighted images with intense contrast uptake on Gadolinium administration and hyperintense on T2-weighted images. Complete spinal MRI was normal and cerebrospinal fluid analysis was negative for neoplasic cells. Sub sequently, The patient was taken up for craniotomy and gross total tumor removal. Intraoperatively, the lesion was highly vascular with solid and cystic components. A total resection of the mass was done. The specimen was composed by giant multinucleated cells with bizarre, pleomorphic, and hyperchromatic nucleus. Large areas of palisading and necrosis and vascular proliferation are also observed with frequent mitosis along with atypia. The postoperative course was uneventful and the CT brain scan was without residual tumor. The child underwent radiotherapy (RT) at a dose of 59.4 Gy in 33 fractions for 6 weeks (Figure 1a, Figure 1b) with concurrent daily Temozolomide (TMZ) Chemotherapy (CH). No residual or recurrent disease was identified in the follow-up MRI. Five months later, during adjuvant TMZ, he presented a focal tonic seizure of the left-sided upper limb with vomiting and neck stiffness suggestive of meningeal irritation. Hydrocephalus without signs of recurrence was found in the CT scan. Lumbar puncture revealed clear cerebrospinal fluid with high protein and without organism in microbial examination. Spinal MRI showed multiple,enhancing, intradural, extramedullary nodules principally at C7–D1 level measuring 11 x 7.5 mm and at D10–D11 level (13 x 5 mm) evoking metastatic lesions and causing severe cord compression with leptomeningeal involvement especially along the filum and conus. Tumor decompression wasn’t possible because it was adherent to spinal cord and dura mater and biopsy revealed an inflammatory infiltrate.The evolution was marked by the occurrence of generalized seizure. He was treated by external ventricular drainage, Depakene and antibiotic therapy. Then, he receivedpalliative spinal axis RT at a dose of 8 Gy. Currently, he remained stable.
Discussion
GCG, previously called monstrocellular BT, mainly occurs in adults, but a decade before that of GBM [6]. It is extremely rare in pediatric patients. Therefore, there are only around 100 reported cases in the literature [7]. The median age is 11 years, but it affects the patients of wide age group ranging from children to young adolescents (4-17 years). A male predominance was noted [7]. This tumor is usually located subcortically in the temporal and parietal lobes. However, other less common sites were described including lateral ventricles, optic chiasm, spinal cord and the cerebellum such our case [8]. Rarely, it is multifocal [9].
GCGs with a clinical and family history of Neurofibromatosis are extremely rare [10] and in some cases, it can be associated with tuberous sclerosis [6]. Their genetic pathway shows p53 mutation in 75 to 89% of the cases without significant EFGR over-expression [11,12]. The clinical presentation is similar to that of common GBM. Signs of raised intracranial pressure are often principal symptoms and patients presenting with headache and vomiting with a shorter preoperative history less than six months in 85%, as in our case [13]. Hemiparesis and seizure episodes were observed as an initial symptom of the tumor. Due to these nonspecific symptoms, the tumor can mimic infections, inflammatory processes and circulatory, immunological disease [6]. So, it is crucial to perform radiological scrutiny to narrow the diagnosis.
On imaging, GCG has no distinguishing features when compared with common GBM [14]. Brain MRI usually reveals a contrast-enhancing heterogeneous mass, with solid and cystic areas, hypointense on T1 and hyperintense on T2 sequences with surrounding edema [6]. MR spectroscopy, diffusion‑weighted imaging and perfusion imaging often offer clues for specific diagnosis of GCG [10].
As mentioned earlier in the case description, the diagnosis is based on histopathological examination: The presence of abundant bizarre multinucleated giant cells means a specific feature that allows to define the GCG as a subtype of GBM [15]. Microscopically, they are highly cellular lesions with nuclei of varying sizes, shapes and numbers, with areas of necrosis, mainly in a pseudopalisading or large ischemic forms. The tumor cells are positive for Glial Fibrillary Acidic Protein (GFAP) which represents the glial origin of the tumor.Immunohistochemistry studies have also shown positivity for S-100, vimentin and alpha-1 antichymotrypsin [6].
The differential diagnosis based on imaging and clinical presentation include low-grade tumors such as ganglioglioma, oligodendroglioma and Pleomorphic Xanthoastrocytoma (PXA) [15]. According to Pant et al, the common features shared by GCG and PXA include numerous giant cells, prominent reticulin stroma, lymphocytic infiltrates and evident circumscription. Supposedly, GCG include, clinically a short history with quicker evolution of seizures, histopathologyshowing numerous bizarre giant cells, frequent atypical mitoses, pseudopalisading necrosis, with immunopositivity for p53 and GFAP and immunonegativity for neuronal markers [14].
Treatment approaches include maximum safe resection along with adjuvant RT [6]. Use of CH has been described as well, although protocols are quite variable [14]. The standard regimen for RT and CH includes fractionated focal irradiation in daily fractions of 2 Gy given 5 days per week for 6 weeks, for a total of 60 Gy and continuous daily TMZ (75 mg per m² of body surface area per day, 7 days per week from the first to the last day of RT), followed by 6 cycles of adjuvant TMZ (150 to 200 mg per m² for 5 days during each 28-day cycle) [15]. The outcome of the GCG patients treated with the more intensive CH regimen was not analyzed [16]. To be noted that surgeryby itself, may offer 32 weeks of mean survival time. RT has proven beneficial, adding 25 weeks to the total mean survival time [14].
The GCG is associated with longer survival times compared to GBM, ranging from 15 months up to 17 years in adults and from 14 months up to 12 years in children [16]. It was linked to a higher degree of complete tumor resection than in GBM patients [7] due to its predominance in locations within the cerebral hemispheres and its less infiltrative behavior such our case [7]. Therefore, the more superficial and localized the tumor, the better the prognosis [6]. Palma et al. Involved the beneficial prognostic influence of limphocytic infiltration in malignant BT and considering that giant monstrous cells are implicated in the host’s enhanced immune response by manifying the antigenic stimulus, that would explain the slower evolution of this tumour [15]. Besides, a study of 18 pediatric patients by Karremann et al. showed no significant difference in median age, male preference, median clinical history, and prognosis between GCG and GBM [7]. Spinal metastasis from primary GBM is extremely rare and occurs relatively late [18,19]. Treatment modalities include surgery for decompression, RT in total dose of 25-40 Gy and intrathecal CH. Because of the diffuse nature of the disease, surgery is generally unsuitable. Therefore, RT is used most commonly. There is no obvious survival advantage of one therapy over the other and the prognostic is poor [18,19].
Conclusion
This case report is focused on the rarest situation of GBM subtype (GCG) which is even few reported, especially in the pediatric population.
Differentiating it from GBM on a clinical and radiological features is difficult because of similar characteristics and is entirely based on histopathological examination with immunochemistry study.
The case presented shows a GCG with non-specific symptoms progressed rapidly over a short time treated by complete removal followed by RT and CH. Unfortunately, the evolution in our patient with spinal leptomeningeal metastases is not habitual and the prognosis is similar to that of common GBM. Future studies with larger cohorts and molecular pathological analyses are still needed to corroborate the findings of the present case report and to establish aconsensual managementof this entity.
References
- Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, et al. CBTRUS statistical report: Primary brain and central nervous system tumorsdiagnosed in the United States in 2006-2010. Neuro Oncol. 2013; 15: ii1-56.
- Brassesco MS, Darrigo LG Jr, Valera ET, Oliveira RS, Yamamoto YA, et al. Giant-cell glioblastoma of childhoodassociated with HIV-1 and JC virus coinfection. Childs Nerv Syst. 2013 ; 29: 1387-1390.
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, et al. Paediatric and adultglioblastoma: multiform (epi)genomicculpritsemerge. Nat Rev Cancer. 2014; 14: 92-107.
- Deb P, Sharma MC, Chander B, Mahapatra AK, Sarkar C, et al. Giant cell glioblastoma multiforme: report of a case with prolongedsurvival and transformation to gliosarcoma. Child’s Nervous System: Chns : Official Journal of the International Society for Pediatric Neurosurgery. 2006; 22: 314-319.
- Palma L, Celli P, Maleci A, Di Lorenzo N, Cantore G. Malignant monstrocellular brain tumours. A study of 42 surgically treated cases. Acta Neurochir (Wien). 1989; 97: 17-25.
- Suraj Shrestha, Sushan Homagain, Akash Raut, Gopal Sedhain, Suraj Bhatta, et al. Giant cell glioblastoma in 6-year-old kid: Report of an unusual case. Clin Case Rep. 2020; 00: 1–5.
- Karremann M, Butenhoff S, Rausche U, Pietsch T, Wolff JE, et al. Pediatric giant cell glioblastoma: New insights into a rare tumor entity. Neuro Oncol. 2009; 11: 323-329
- Queiroz LS, Faria AV, Zanardi VA, Netto JR. Lipidizedgiant-cell glioblastoma of cerebellum. Clin Neuropathol. 2005; 24: 262-266.
- Parekh HC, Sharma RR, Prabhu SS, Keogh AJ, Lynch PJ, et al. Multifocal giant cell glioblastoma: case report. SurgNeurol. 1993; 40: 151-154.
- Pant I, Nazir W, Ujjawal V, Chaturvedi S. Giant Cell Glioblastoma in a Child with Clinical and Family History of Neurofibromatosis. Asian J Neurosurg. 2017; 12: 779-782.
- Peraud A, Watanabe K, Schwechheimer K, Yonekawa Y, Kleihues P, Genetic profile of the giant cell glioblastoma. Lab Invest. 1999;79:123-129.
- Peraud A, Watanabe K, Plate KH, Yonekawa Y, Kleihues P, et al. P 53 mutations versus EGF receptor expression in giant cell glioblastomas. J Neuropathol Exp Neurol 1997; 56: 1236-1241.
- Georgiu C, MihuŢ E, Raus I, Mirescu ŞC, Szabo L, et al. Pediatric glioblastoma with giantcells and “supratentorial” primitive neuroectodermal component - case report and review of the literature. Rom J MorpholEmbryol. 2015; 56: 1165-1171.
- Jain SK, Sundar IV, Sinha VD, Sharma V, Bhasme V, et al. Giant cell glioblastoma in a child: A rare case report. Asian J Neurosurg. 2012; 7: 144-146.
- De Prada I, Cordobés F, Azorín D, Contra T, Colmenero I, et al. ediatric giant cell glioblastoma: A case report and review of the literature. Childs Nerv Syst. 2006; 22: 285-289.
- Zipp L, Schwartz KM, Hewer E, Yu Y, Stippich C, et al. Magnetic resonance imaging and computedtomographyfindings in pediatricgiant cell glioblastoma. Clin Neuroradiol. 2012 ; 22: 359-363.
- Sharma D, Gupta A, Dhillon GS, Chhabra SS. Late on set leptom eningeal and whole spine metastasis from supratentorial Glioblastoma multiforme: An un common manifestation of a common tumor. J Craniovertebr Junction Spine. 2016; 7: 118-120.
- Witoon panich P, Bamrungrak K, Jinawath A, Wongwaisayawan S, Phudhichareonrat S, lioblastoma multiforme at the corpus callosum with spinal leptomeningeal metastasis. Clin Neurol Neurosurg. 2011; 113: 407-410.