
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Moyamoya disease: A case report in a 10 years old Sudanese girl
Amjed Abdu1; Thowiba Mohammed2; Mustafa Ahmed3; Basharia Abdalla4; Sufian Khalid5*
1Assistant Professor, Department of Pediatrics, Faculty of Medicine, Nile Valley University, Sudan.
2Houseman Ship at ATBRA Teaching Hospital, River Nile State, Sudan.
3Teaching Assistant, Department of skills Lab and Problem Based Learning, Faculty of Medicine, Ahfad University for Women, Sudan.
4Houseman Ship at ATBRA Teaching Hospital, River Nile State, Sudan.
5Professor of Internal Medicine, Faculty of Medicine, Nile Valley University, Sudan.
*Corresponding Author : Sufian Khalid Mohammed Noor
Professor of Internal medicine, Faculty of Medicine, Nile Valley University, Sudan.
Email: sufiankhaid@yahoo.com
Received : Apr 08, 2023
Accepted : Jun 20, 2023
Published : Jun 27, 2023
Archived : www.jcimcr.org
Copyright : © Noor SKM (2023).
Abstract
Background: Moyamoya Disease (MMD) is a rare pathology caused by a progressive unilateral or bilateral stenosis of the terminal portion of the internal carotid artery, leading to thedevelopment of collateral vessels.
Case presentation: We report a rare case of a 10 years old girl presented with sudden onset of left side weakness not associated with headache or vomiting. The diagnosis of MMD was established when the cerebral angiogram revealed moderate proximal right MCA stenosis together with a plastic A1. The left ICA showed T occlusion at bifurcation with very strong collaterals.
Conclusion: This case highlights the importance of considering moyamoya disease to be one of the classic etiologies of acute ischemic strokes in children from North Africa. It also emphasizes the rare presentation among the African population and the use of neurovascular imaging techniques to facilitate diagnosis of moyamoya disease.
Citation: Abdu A, Mohammed T, Ahmed M, Abdalla B, Noor SKM. Moyamoya disease: A case report in a 10 years old Sudanese girl. J Clin Images Med Case Rep. 2023; 4(6): 2474.
Introduction
Moyamoya disease is a chronic, progressive occlusion of the circle of Willis arteries that leads to the development of characteristic collateral vessels evidence by imaging, particularly cerebral angiography [1]. The disorder initially involves the intracranial portion of the internal carotid arteries and progresses to involve the middle, anterior and posterior cerebral arteries. Collateral vascular network developed due to compensatory dilatation of lenticulostriate and thalamostriate arteries. Angiographic study of this collateral vascular network gives rise to characteristics appearance such as a pu of cigarette smoke. ¬e Japanese term “MOYAMOYA” means “something hazy like a pu of cigarette smoke [2,3].
Moyamoya disease was first described in Japan by Takeuchiand Shimizu in 1957. Oughe disease is most common in Japan, many subsequent cases have been reported elsewhere Asia, Europe and North America. In 1988 the Japanese Ministry of Health and Welfare defined primary MMD as an idiopathic bilateral stenosis of arteries of the circle of Willis with collateral vascular networks, demonstrated on angiography. Recent studies indicate that the amount of collateral vessels at the base of the skull serve as markers of diseaseseverity and degree of progression [4]. The gold standard in the current diagnosis of Moyamoya disease is cerebral angiography. As it is an invasive exam, wecan use other methods such as angiotomography and MR angiography, which are non-invasive exams. Currently, there is no specific treatment to prevent the progression of Moyamoya disease. However, the procedure predilection for patients with ischemic and hemorrhagic strokes is surgical revascularization (extracranial-intracranial bypass) [5,6].
Case presentation
A 10 years old girl of consanguineous parents presented complain of left side weakness and right loin pain for 2 days. Here parents stated that she developed sudden left side hemiplegia to extent that she unable to stand or walk. After this episode she gradually improved in standing & walking but weakness persist that made her walking difficult There was no history of fever, Loss of consiousness, vomiting, or aphasia along with this illness and no history of radiation exposure or head trauma mentioned before this. No family history of similar condition. Had past history of appendectomy, not on long term medications. On examination she has no dysmorphic features, vital signs within normal range, no skin change. Neurological examinations revealed that she had normal tone, power 4l5 on left side along with hyperreflexia, exaggrarated ankle clonus and positive Babinski sign. Other CNS examinations were normal. Investigations requested include complete blood count, random blood glucose, urine analysis were normal abdominal ultrasound revealed right renal stone. MRI brain with contrast revealed: Bilateral signal changes involving both temporal lobes with partial involvement of adjacent gyri of both frontal lobes may represent encephalitis. The acute infract involving the territory of MCA could be a complication of this encephalitis or represent separate entity so we have periventricular leukomalacia more extensive seen on left side, this could be a result of previous hypoxic event involving branches of this left MCA. The bilateral involvement of deep white matter of both cerebellar hemispheres with associated cortical multiple ischemic foci could be a result of posterior circulation insuffiency.
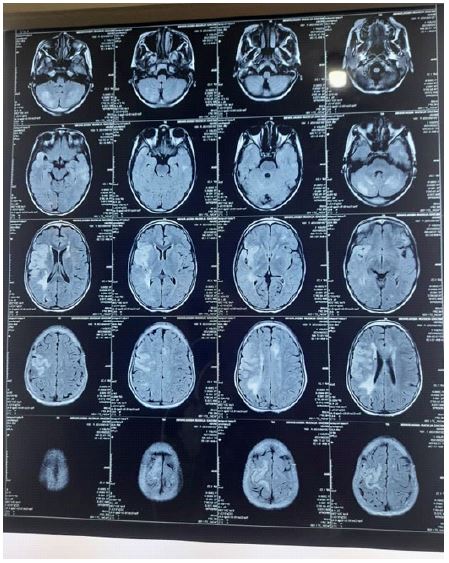
Other investigations was done cerebral angiogram and revealed moderate proximal right MCA together with a plastic A1. The left ICA showed T occlusion at distal bifurcation with very strong collateral. Normal vertebra-basilar system. Features consist with vasculitis.

Discussion
Compared with adults, acute stroke is an infrequent disease of pediatric patients. In fact, the reported incidence of childhood stroke has increased in the last 20 years according to population studies, most likely related to improvements in neurological techniques [7]. There are numerous causes of stroke in childhood, These include congenital heart disease, sickle disease, immune disorders, clotting disorders, and head and neck trauma. However, MMD should be included in the differential for a stroke in a child. In fact, MMD represents one of the most severe arteriopathies and accounts for approximately one fifth of identified cerebral arteriopathies in children presenting with acute stroke and is diagnosed in up to 20% of cases of childhood Acute Ischemic Stroke (AIS) [8]. The condition is common In Japan and East Asia, where familial cases are also clearly recognized. In Japan, the annual prevalence and incidence have been estimated to be 3.16–10.5 and 0.35–0.94 per 100,000 [9]. The female to male ratio has been shown to be 1.8-2.2 (female predominance) 10.The familial form accounts for 10–15% [11].
Moyamoya is categorized as MMD when there is no underlying etiology or association and as moyamoya syndrome if an underlying etiology or association of other conditions is recognized, including trisomy [21], Down syndrome, neurofibromatosis type 1, and cranial irradiation. In our patient, no association of any other systemic disorder was described. Furthermore, most general medical conditions were excluded by normal blood, urine studies, and negative history of fever or trauma. A normal physical examination ruled out increased intracranial pressure as well as meningitis. Pathological changes in moyamoya patients include intimal thickening with brous tissue, abnormalities of internal lamina elastica, variable lipid deposition and virtual absence of inflammatory reaction in the blood vessel [12,13].
Clinically, the presentation of patients with moyamoya disease may include seizures, transient ischemic attacks, ischemic strokes, and hemorrhagic strokes [14-16]. Visual decits, speech disturbance, migrane like headache, intellectual deterioration, cranial nerve palsies, and disturbance of gait can also be evident [17,18]. Our patient presented with ishchemic stroke. Conventional angiogram or Digital Subtraction Angiogram (DSA) is the investigation of choice. MRA has a reliable diagonostic modality for MMD [19]. Treatment in the acute phase is symptomatic with the goal of maintaining cerebro-vascular perfusion and function. Anticoagulant and antiplatelet agents have shown no remarkable benet [20]. The same lack of obvious ecacy has been described for corticosteroids in moyamoya disease [21]. McLean et al, elucidated the use of verapamil hydrochloride to curtail the ischemic symptoms associated with moyamoyadisease [22], Surgical treatment modalities have been used to manage the hemorrhagic and ischemic consequences of moyamoya disease [23-25,27]. Direct revascularization techniques, which are typically used in adults, include the supercial temporal artery to middle cerebral artery bypass or the middle meningeal artery to middle cerebral artery bypass are commonly applied in children [26,27].
Conclusion
Although Moyamoya is predominat in Japanese population but should not overlooked in other population.The patient who will fulll the clinical characteristics, MR angiogram should be done to diagnose Moyamoya disease.
References
- Dr Gopen Kumar Kundu, Associate Professor, Institute of Paediatric Neuro disorder and Autism (IPNA), Bangabandhu Sheikh Mujib Medical University Dhaka. Email: gopen.kundu@gmail.com
- Dr Md Benzamin, Resident, Department of Paediatric Gastroenterology & Nutrition, Bangabandhu Sheikh Mujib Medical University, Dhaka.
- Dr Subrota Kumar Roy, Assistant Professor, Department of Paediatrics, Dhaka Medical College, Dhaka.
- Zhao M, Zhang D, Wang S, Zhang Y, Deng X, Zhao J, et al. The Collateral Circulation in Moyamoya Disease: A Single-Center Experience in140 Pediatric Patients. Pediatr Neurol. 2017; 77: 78-83.
- Bang OY, Fujimura M, Kim SK. The Pathophysiology of Moyamoya Disease: An Update. J Stroke. 2016; 18: 12-20.
- Franco CMR, Fukujima MM, Oliveira RMC, Gabbai AA, et al. Moyamoya disease: Report of three cases in Brazilian patients.ArqNeuropsiquiatr. 1999; 57: 371–376.
- Mirsky DM, Beslow LA, Amlie-Lefond C, Krishnan P, Laughlin S, et al. International Paediatric Stroke Study Neuroimaging Consortium, Paediatric Stroke Neuroimaging Consortium. Pathways for Neuroimaging of Childhood Stroke. Pediatr Neurol. 2017; 69: 11–23.
- Amlie-Lefond C, Ellenbogen RG. Factors associated with the presentation of moyamoya in childhood. J Stroke Cerebrovasc Dis. 2015; 24: 1204–1210.
- Kuriyama S, Kusaka Y, Fujimura M, Wakai K, Tamakoshi A, HashimotoS, et al. Prevalence and clinicoepidemiological features of moyamoya disease inJapan: findings from a nationwide epidemiological survey. Stroke. 2008; 39: 42–47.
- Kim SK, Cho BK, Phi JH, Lee JY, Chae JH, Kim KJ, et al. Paediatricmoyamoya disease. An analysis of 410 consecutive cases. Ann Neurol. 2010; 68: 92–101.
- Takanashi J. Moyamoya disease in children. Brain and Development. 2011; 33: 229–233.
- Yilmaz EY, Pritz MB, Bruno A, Lopez-Yunez A, BillerJ. Moyamoya: Indiana University Medical Centerexperience. Arch Neurol. 2001; 58: 1274-1277.
- Inoue TK, Ikezaki K, Sasazuki T, Matsushima T, FukuiM. Analysis of classII genes of human leukocyteantigen in patients with moyamoya disease. Clin Neurol Neurosurg. 1997; 99: S234-S237.
- Bertora P, Lovati C, Gambaro P, Vicenzi A, Rosa S, Osio M, et al. Moyamoya disease in a member of the Roma gypsy community. Eur Neurol. 2008: 59: 274-275.
- Wakai K, Tamakoshi A, Ikezaki, Fukui M, Kawamura T, Aoki R, et al. Epidemiological features of moyamoyadisease in Japan: ndings from a nationwide survey. Clin Neurol Neurosurg. 1997; 99: S1-S5.
- Fukui M. Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya disease)of the Ministry of Health and Welfare, Japan. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis. Clin Neurol Neurosurg. 1997; 99: S238-S240.
- Kitamura K, Fukui M, Oka K. Moyamoya disease. In: Handbook of Clinical Neurology. Amsterdam, e Netherlands: Elsevier; 1989; 2: 293-306.
- Hoare AM, Keogh AJ. Cerebrovascular moyamoya disease. Br Med J.1974; 1: 430-432.
- Sakurai K, Horiuchi Y, Ikeda H, Ikezaki K, Yoshimoto T, Fukui M, et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004; 49: 278-281.
- Takanashi J, Sugita K, Honda A, Numi H. Moyamoyasyndrome in a patient with Down syndrome presenting with chorea. Pediat. Neurol. 1993; 9: 396-398.
- Spittler JF, Smektala K. Pharmacotherapy inmoyamoya disease. HokkaidoIgaku Zasshi. 1990; 65: 235-240.
- McLean MJ, Gebarski SS, vanderSpek AF, Goldstein GW. Response of moyamoya disease to verapamil. Lancet. 1985; 1: 162-163.
- Uchino K, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington State and California. Neurology. 2005; 65: 956-958.
- Yilmaz EY, Pritz MB, Bruno A, Lopez-Yunez A, Biller J, et al. Moyamoya: Indiana University Medical Center experience. Arch Neurol. 2001; 58: 1274-1277.
- Chiu D, Shedden P, Bratina P, Grotta JC. Clinicalfeatures of moyamoya disease in the United States.Stroke. 1998; 29: 1347-1351.
- Kuroda S, Ishikawa T, Houkin K, Nanba R, Hokari M, Iwasaki Y, et al. Incidence and clinical features of disease. Progression in adult moyamoya disease. Stroke. 2005; 36: 2148-2153.
- Oya S, Tsutsumi K, Ueki K. Adult-onset moyamoyadisease with repetitive ischemic attacks successfullytreated by supercial temporal-middle cerebral artery bypass - case report. Neurol Med Chir. 2003; 43: 138-141.