
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
A rare case of Rodriguez syndrome caused by a SF3B4 gene deletion
Anupama Chundu1*#; Joyce Hsiou2,3#; Yona Nicolau1; Rob Edwards4; Michelle Bosworth5; Roger Schultz6; Bo Hong5,6; Virginia Kimonis2,4,7*
1Division of Neonatology, Department of Pediatrics, University of California Irvine, USA.
2Division of Genetics and Genomic Medicine, Department of Pediatrics, University of California Irvine, USA.
3Western University of Health Sciences, Pomona, California, USA.
4Department of Pathology, University of California Irvine, USA.
5ARUP Laboratories, Salt Lake City, UT, USA.
6Department of Pathology, University of Utah, Salt Lake City, UT, USA.
7Department of Neurology, University of California Irvine, USA.
*Corresponding Author : Anupama Chundu
Department of Pediatrics, University of California Irvine Medical Center, 101 The City Drive South, Bldg 56, Suite 600, Orange, CA 92868-3298, USA.
Tel: 714-456-6920 & 714-456-7658;
Email: Achundu@hs.uci.edu
Virginia E Kimonis
Department of Pediatrics, University of California Irvine Medical Center, 101 the City Drive South, ZC4482, Orange CA 92868, USA.
Tel: 714-456-5791 & 714- 456-5330;
Email: vkimonis@uci.edu
Received : Jul 03, 2023
Accepted : Jul 19, 2023
Published : Jul 26, 2023
Archived : www.jcimcr.org
Copyright : © Anupama C & Virginia K (2023).
Abstract
The acrofacial dysostoses are a genetically heterogeneous group of inherited disorders characterized by craniofacial and limb abnormalities. Nager and Rodriguez syndromes, posited as distinct subtypes, have recently been described as a phenotypic spectrum associated with haploinsufficiency of the SF3B4 gene. A 27-year-old primigravida whose pregnancy was complicated by polyhydramnios and fetal anomalies that consisted of borderline ventriculomegaly, micrognathia, hypotelorism, microphthalmia, arthrogryposis, talipes, and low set ears delivered at 31 weeks 2 days gestation. Numerous intubation attempts failed due to micrognathia, and the baby only survived a few hours. The cause of death was determined to be respiratory insufficiency. Clinical features were suggestive of Nager/Rodriguez syndrome, and cytogenomic SNP microarray showed an interstitial deletion involving chromosome 1 within 1q21.2q21.3. This region includes 27 protein coding genes; most importantly the SF3B4 gene was deleted. Only two cases of a SF3B4 whole gene deletion causing Rodriguez syndrome have been reported, one in a fetus at 12 weeks gestation and another at 13 weeks gestation [1,2].
Citation: Chundu A, Hsiou J, Nicolau Y, Edwards R, Kimonis V, et al. A rare case of Rodriguez syndrome caused by a SF3B4 gene deletion. J Clin Images Med Case Rep. 2023; 4(7): 2516.
Introduction
Acrofacial dysostosis is a group of disorders characterized by distinctive craniofacial malformations, especially involving severe mandibular hypoplasia and limb defects.
Multiple subtypes have been described which vary in specific pattern of anomalies, severity, and inheritance pattern. Nager syndrome includes preaxial limb anomalies and is most often associated with SF3B4 mutations, which encodes a spliceosome protein. A variant of Nager is lethal in the perinatal period, has notable specific differences in presentation and has been designated Rodriguez syndrome [1]. Variants in SF3B4 have been identified in patients with Rodriguez syndrome [3]. We report a unique case of Rodriguez syndrome caused by an interstitial deletion involving chromosome 1q21.2q21.3, this region including the SF3B4 gene.
Case presentation
The baby was born to a 27-year-old primigravida mother whose pregnancy was complicated by polyhydramnios and multiple fetal anomalies. There were no reports of potential teratogenic exposure. The prenatal ultrasound at 30 weeks gestation revealed borderline ventriculomegaly, micrognathia, hypotelorism, microphthalmia, arthrogryposis, talipes, and low set ears. In view of the fetal micrognathia and limb contractures the diagnosis considered prenatally was Freeman-Sheldon syndrome. The ethnic background is Northern European and Native American in the mother and Japanese in the father. The family history was significant for hip dysplasia in the mother and achondroplasia in a maternal cousin. Dementia was reported in the paternal grandparents and Down syndrome in a paternal cousin.
Following preterm premature rupture of membranes and labor, the baby was born at 31 weeks and 2 days gestation by emergency cesarean section for a footling breech presentation.At delivery, baby was apneic and required resuscitation. Apgar scores were 4 at 1 minute of life, 7 at 5 minutes of life, and 7 at 10 minutes of life. Due to severe micrognathia, numerous in-tubation attempts were unsuccessful. A laryngeal mask airway was placed and infant was successfully ventilated via bag and mask with 100% O2 until the baby was taken to the operating room (OR) for a tracheostomy. Upon arriving at the operating room, baby’s heart rate dropped to less than 60 beats per minute and she required chest compressions, numerous rounds of epinephrine, normal saline volume boluses, and was successfully intubated. Decisions were made to defer tracheostomy and plan a mandibular distraction once the baby was more stable. The infant continued to be bradycardic and hypotensive requiring fluid resuscitation, vasopressors, hydrocortisone, high frequency oscillatory ventilation and inhaled nitric oxide for severe hypoxia with concerns for lung hypoplasia and pulmonary hypertension. Despite maximal cardiorespiratory support she succumbed from respiratory failure.
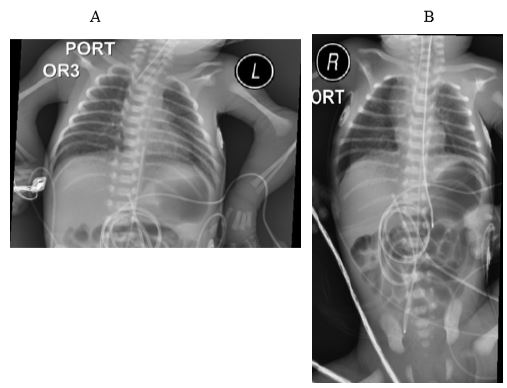
The baby was noted to have multiple congenital anomalies including severe micrognathia with normal tongue size, contractures, and low set ears with bifid and unattached helices, ear canals anteriorly displaced from the helices. There were flexion and rotation contractures of bilateral elbows, and ankles (arthrogryposis) with overlying ecchymoses, absent thumbs bilaterally, and a sacral dimple. The baby’s clinical features were also remarkable for diffuse bruising across the face and chest, bruising of all four extremities, soft swelling of the right parietal scalp, and a nuchal skin fold. Baby’s birth weight was 1225 grams (18.9%), length was 39 cm (31.9%) and head circumference was 28 cm (46.1%). Facial features included cloudy corneas, frontal bossing, trigonocephaly, midface hypoplasia, and anteverted nares. The limbs were significantly shortened with joint contractures. Radiographic studies of the chest revealed hypoplastic ribs, radio-ulnar synostosis, shoulder girdle and pelvic hypoplasia, and phocomelia of upper limbs as the most, but no other significant anomalies were noted (Figure 1A,B). Parents did not provide permission to publish the images showing the craniofacial and limb anomalies.
On autopsy, baby had pulmonary hypoplasia, with the right lung weighing 9.66 g and the left lung weighing 9.3 g (expected 28.5 + 13.2 g). Pulmonary isomerism was also present with both lungs being bilobed. Peritracheal hemorrhage was noted likely secondary to the numerous intubation attempts. There were subserosal and luminal hemorrhage in the gastrointestinal tract involving 14 inches of mid and distal small intestine. Cardiac findings included a patent foramen ovale and patent ductus arteriosus. There was a 9 x 7 cm subgaleal hemorrhage but otherwise the brain architecture and pathology were normal.
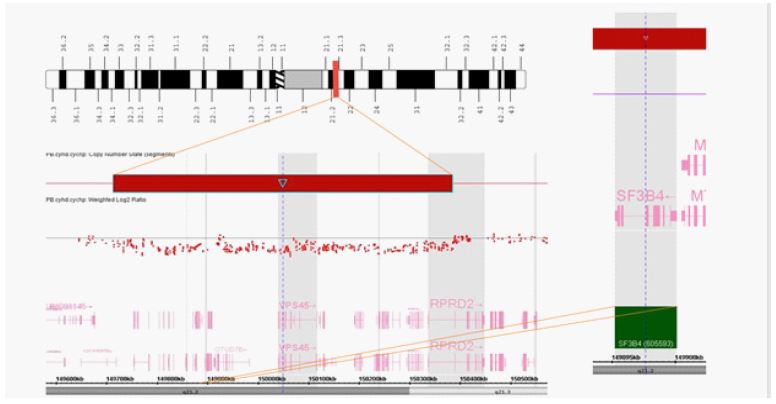
The baby’s features were most suggestive of a severe variant of Nager designated Rodriguez acrofacial dysostosis, allelic disorders associated with SF3B4 gene variants resulting in haploinsufficiency. A genomic SNP microarray (GMA) was performed on a peripheral blood specimen using a CytoScan HDTM platform and analyzed using Chromosome Analysis Suite software (ChAS) (Applied Biosystems/Thermo Fisher Scientific, Santa Clara, CA). The GMA analysis revealed an interstitial deletion of chromosome 1q21.2q21.3. This region includes 27 protein coding genes, most importantly the SF3B4 gene (Figure 2). The other genes in the region were not known to be linked to congenital anomalies when deleted. Maternal SNP microarray performed on a peripheral blood specimen was normal. The father did not pursue testing, however it was thought unlikely that he would be positive for this interstitial deletion.
Discussion
The acrofacial dysostoses are a genetically heterogeneous group of inherited disorders characterized by craniofacial and limb abnormalities. Nager syndrome is characterized by malar hypoplasia, down slanting palpebral fissures, mandibular hypoplasia, microtia and/or malformed external ears resulting in blind ending or absent external ears leading to conductive hearing loss, and characteristically radial limb abnormalities, most commonly underdevelopment or absence of the thumbs and radius as well as radioulnar synostosis [4]. Less than 100 cases have been reported in the literature and although it is rare, it is the most common form of acrofacial dysostosis. The molecular basis of Nager syndrome was first identified as resulting from haploinsufficiency of the mRNA spliceosomal protein splicing factor 3B, subunit 4 (SF3B4) gene [5].
Rodriguez syndrome has many shared features with Nager syndrome such as mandibular hypoplasia, microtia, cleft palate, and hand anomalies. However, reported features unique to the more severe Rodriguez syndrome include severe facial involvement resembling mandibulofacial dysostosis, small stature, phocomelia, oligodactyly of the upper limbs which is predominantly preaxial, severe hypoplasia of the shoulder and pelvic girdles, severe internal organ anomalies such as arrhinencephaly, abnormal long lobulation and congenital cardiac defects [6]. Rodriguez syndrome has been reported to be caused by severe frameshift mutations in the SF3B4 gene [6]. Perinatal mortality is reported in 20% and is related to respiratory distress secondary to micrognathia and palatal abnormalities. Our case has many of the severe manifestations of Rodriguez syndrome.
Nager and Rodriguez syndromes are allelic disorders commonly due to haploinsufficiency of the mRNA spliceosomal protein splicing factor 3B, subunit 4 (SF3B4) gene [5]. The mutations lead to reduced SF3B4 synthesis and defects in mRNA splicing, primarily exon skipping, thus important in gene expression pathways [7]. They also lead to reduced expression in growth plate chondrocytes of target genes which are known to be important for skeletal development [7]. The mutation is usually a single base insertion causing frameshifts, however in the case of our baby, the SNP microarray showed a deletion of the SF3B4 gene. Only two other cases of a SF3B4 whole gene deletion causing Rodriguez syndrome have been reported in the literature [1,2]. The fetal findings reported in the paper by Lund et. al (2016) are micrognathia, malformed wrists, bilateral club foot, and short long bones [1]. Drozniewska et al. (2020), reported a fetus with micrognathia, bilateral club foot, short long bones, and cleft lip [2]. These case reports and our case with many of the same findings thus suggests that SF3B4 haploinsufficiency is the mechanism also for the more severe Rodriguez syndrome. For those variants that were common to both Nager and Rodriguez syndromes, we cannot rule out another mechanism such as a mutation in the promoter or enhancer region of the gene or an undetected second variant resulting in a more severe manifestation.
Acknowledgements: We thank the family for giving permis-sion to publish this case and the health care providers involved in her care. We thank Mabel Tang for her expert administrative assistance.
References
- Lund IC, Vestergaard EM, Christensen R, Uldbjerg N, Becher N. Prenatal diagnosis of Nager syndrome in a 12-week-old fetus with a whole gene deletion of SF3B4 by chromosomal microarray. Eur J Med Genet. 2016; 59: 48-51.
- Drozniewska M, Kilby MD, Vogt J, et al. Second-trimester prenatal diagnosis of Nager syndrome with a deletion including SF3B4 detected by chromosomal microarray. Clinical Case Reports. 2020; 8: 508-511.
- Drivas TG, Taylor JA, Zackai EH. The final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature. Am J Med Genet A. 2019; 179: 1063-1068.
- Czeschik JC, Voigt C, Alanay Y, et al. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet. 2013; 132: 885‐898.
- Petit F, Escande F, Jourdain AS, Porchet N, Amiel J, et al. Nager syndrome: Confirmation of SF3B4 haploinsufficiency as the major cause. Clinical genetics. 2014; 86: 246-251.
- Dimitrov B, Balikova I, Jekova N, Vakrilova L, Fryns JP, et al. Acrofacial dysostosis type Rodriguez. Am J Med Genet A. 2005; 135: 81-85.
- Marques, Felipe, et al. Altered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis. PLoS genetics. 2016; 12: e1006307.