
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Kufor-Rakeb syndrome in two Iranian brothers due to different and novel mutations
Reza Mosaddeghi-Heris*; Nasrin Forghani; Sorayya Sarvi
Neurosciences Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
*Corresponding Author : Reza Mosaddeghi Heris
Neurosciences Research Center, Tabriz University of Medical Sciences, Tabriz, Iran.
Ph: +98-9149101628;
Email: rezamosaddeghi1375@gmail.com
ORCID ID: 0000-0002-3353-1102
Received : Jun 05, 2023
Accepted : Aug 03, 2023
Published : Aug 10, 2023
Archived : www.jcimcr.org
Copyright : © Mosaddeghi-Heris R (2023).
Abstract
Kufor-Rakeb Syndrome (KRS) is an uncommon autosomal recessive neurological condition that frequently has clinical symptoms including supranuclear gaze palsy, dementia, and generalized brain atrophy. Patients with KRS have been shown to carry mutations in the PARK9 locus-related gene, ATP13A2. Here, we present the KRS cases of two Iranian brothers who also have a new mutation in ATP13A2 in addition to a pathogenic mutation.
Keywords: KuforRakeb Syndrome; ATPase type 13A2 protein; Hereditary spastic paraplegia; Novel mutation.
Abbreviations: KRS: Kufor-Rakeb Syndrome; PD: Parkinson’s Disease; HSP: Hereditary Spastic Paraplegia; WES: Whole Exome Sequencing; DTR: Deep Tendon Reflex; MF: Muscle Force; EMG: Electromyography; NCV: Nerve Conduction Studies; CMAPs: Compound Muscle Action Potentials.
Citation: Mosaddeghi-Heris R, Forghani N, Sarvi S. Kufor-Rakeb syndrome in two Iranian brothers due to different and novel mutations. J Clin Images Med Case Rep. 2023; 4(8): 2537.
Intoduction
Kufor-Rakeb Syndrome (KRS) is an autosomal recessive form of Parkinson’s Disease (PD) characterized by juvenile-onset [1]. There may be pyramidal signs, dysarthria, dysphagia, and cognitive difficulties in adolescence in patients with this disease [2]. It has been found that KRS patients carry mutations in the ATP13A2 gene (chromosome 1p36) associated with the PARK9 locus. This gene encodes a P5B-type lysosomal ATPase that shares functional domains with other P-type ATPases [1]. There is impaired Mn2+ and Zn2+ metabolism, mitochondrial homeostasis, and lysosomal function associated with mutations of ATP13A2 [3]. This paper reports two Iranian brothers who present with KRS and have a known pathogenic mutation as well as a novel mutation for ATP13A2.
Case presentation
lower extremities weakness and gait impairment since seven years ago. In the first year of the disease, he was diagnosed with Hereditary spastic paraplegia (HSP) due to physical examination and Ears of the lynx sign (Figure 1) and atrophic changes in the cerebellum (Figure 2) on brain MRI. But he refused to perform the Whole Exome Sequencing (WES) exam. Furthermore, he has a 30 y/o brother (B) with the same clinical symptoms, who hasn’t sought a medical diagnosis. Their parents were non-consanguineal. He came to the neurology clinic due to an exacerbation of symptoms. There was no sign of skeletal deformity, muscular atrophy, or involvement of the sphincter in the clinical examination. The gait was spastic, plantar reflexes were extensor on both sides, and Deep Tendon Reflexes (DTRs) were 3+ in the lower limbs and 2+ in the upper limbs. Muscle Forces (MFs) were 4/5 in the proximal and 3/5 in the distal of the lower extremities, and 5/5 in the upper limbs. His brother had a spastic gait with brisk DTRs in lower limbs and double extensor plantar reflexes and diminished MFs in lower limbs, 3/5 in proximal and 3/5 in distal.
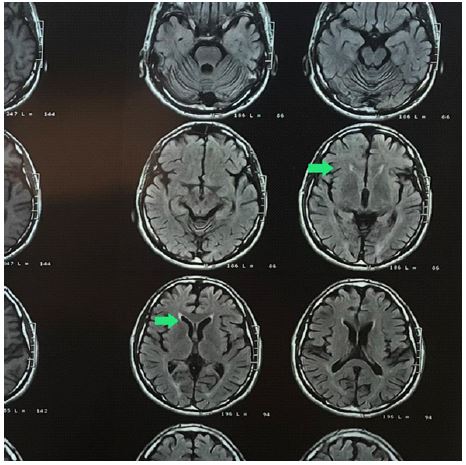

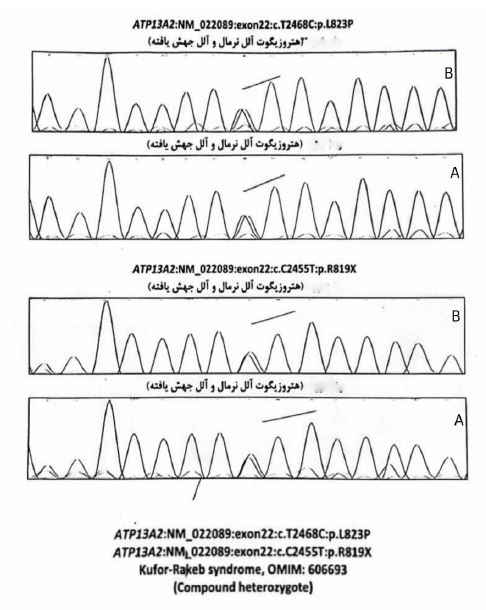
His previous EMG/ NCV suggested an upper motor neuron process. The results of the WES exam showed both of them were compound heterozygous for the ATP13A2: NM gene for both of them (Figure 3). One of the reported mutations was previously reported as a pathogenic mutation (p.R819X), and the other mutation (p.L823P) is novel. As a result of their genetic consultation, their family is carrying this mutation, so their children will be prone to it (25%). Hence, we advised them to seek genetic consultation before family planning. Also, to improve the patient’s condition, occupational therapy was recommended, which the patient refused.
Discussion
We reported 2 Iranian brothers with compound heterozygous for ATP13A2 who presented with KRS with a known pathogenic mutation and a novel mutation. Although the parents of the patients were not available to determine the genotype, and their father is also deceased, it is highly probable that both parents had one of the different mutations, and the children inherited them.
Patients with KRS carry homozygous or compound heterozygous loss-of-function mutations consistent with their autosomal recessive inheritance pattern. In addition to identifying ATP13A2 as a disease-causing gene, the identification of ATP13A2 resulted in laboratory studies of its molecular function, as well as defining the pathophysiological mechanisms that contribute to the clinical phenotype [3]. From various disease models, what could be concluded at this time is that ATP13A2 has a function that is associated with Mn2+ and Zn2+ metabolism [4,5], mitochondrial bioenergetics [6-8], and the autophagy–lysosome pathway [6]. Furthermore, ATP13A2 is also believed to modulate α-synuclein metabolism, which is considered to be one of the main components of Lewy bodies [9]. In spite of its more complicated clinical presentation, the discovery of this monogenic form of PD has led to numerous studies investigating whether this gene is involved in the development of sporadic PD as well. It has been discovered that a number of single heterozygous ATP13A2 mutations are much more common in early-onset PD patients than in healthy controls, indicating that these mutations may operate as a risk factor or age-of-on set modifier for PD [10].
The nerve conduction studies of some previously reported cases revealed a decrease in motor amplitudes in the lower limbs, along with evidence of impaired central conduction on somatosensory evoked potentials and involvement of the corticospinal tract on motor evoked potentials [3]. Likewise, our case had mildly decreased amplitude bilaterally in tibial Compound Muscle Action Potentials (CMAPs).
Acknowledgments: In this study, the authors would like to express their gratitude to the patients and families that consented to participate in the study. It is confirmed that this work was not subject to institutional review board approval.
References
- JS Park and CM Sue. Hereditary Parkinsonism-Associated Genetic Variations in PARK 9 Locus Lead to Functional Impairment of ATPase Type 13A2, (in eng), Curr Protein Pept Sci. 2017; 18: 7725-7732.
- DR Williams, A Hadeed, AS al-Din, AL Wreikat, AJ Lees, et al. Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia, (in eng), Mov Disord. 2005; 20: 1264-1271.
- JS Park, NF Blair, CM Sue. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Movement Disorders. 2015; 30: 770-779.
- AD Gitler, et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity, Nature genetics. 2009; 41: 308-315.
- SM Kong, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes, Human molecular genetics. 2014; 23: 2816-2833.
- AM Gusdon, J Zhu, B Van Houten, CT. Chu, et al. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy, Neurobiology of disease. 2012; 45: 962-972.
- D Ramonet, et al. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity, Human molecular genetics. 2012; 21: 1725-1743.
- A Grünewald, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome, Neurobiology of aging. 2012; 33: 1843. e1-1843. e7.
- M Usenovic, E Tresse, JR Mazzulli, JP Taylor, D Krainc, et al. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity, Journal of Neuroscience. 2012; 32: 4240-4246.
- A. Di Fonzo, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease, Neurology. 2007; 68: 1557-1562.