
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Series - Open Access, Volume 4
Primary cutaneous myoepithelial carcinoma: A report of two cases with literature review
Manjusha Mullappli P1*; Sangeetha Nayanar1; Divya Radhakrishnan1; Nizamudheen Pareekuty2; Satheesh Babu3
1Department of Onco-Pathology, Malabar Cancer Centre, Thalassery, Kerala, India.
2Department of Surgical Oncology, Malabar Cancer Centre, Thalassery, Kerala, India.
3Department of Imageology, Malabar Cancer Centre, Thalassery, Kerala, India.
*Corresponding Author : Manjusha Mullappli P
Department of Onco-Pathology, Malabar Cancer Centre, Thalassery, Kerala.
Email: manjusha91@gmail.com
Received : Aug 04, 2023
Accepted : Aug 28, 2023
Published : Sep 04, 2023
Archived : www.jcimcr.org
Copyright : © Mullappli PM (2023).
Citation: Mullappli PM, Nayanar S, Radhakrishnan D, Pareekuty N, Babu S. Primary cutaneous myoepithelial carcinoma: A report of two cases with literature review. J Clin Images Med Case Rep. 2023; 4(9): 2575.
Background
Myoepithelial tumors encompass a spectrum of neoplasms demonstrating myoepithelial differentiation. While originally well-described in salivary glands, these tumors have been increasingly reported in bone, soft tissue, and cutaneous tissues [1]. Although the malignant counterpart of myoepithelial lesions is even rarer in salivary glands, it tends to be the majority of cases in soft tissue/skin. Precise identification and categorization of lesions with myoepithelial differentiation necessitate judicious usage of ancillary testing, especially immunohistochemistry. However, literature on this entity remains limited, with cases showing equal predilection for males and females, typically presenting in middle age, and frequently located in the hip girdle and extremities [1-6].
Histologically, myoepithelial lesions exhibit divergent architecture and are composed of epithelioid, round, spindle, rhabdoid, or plasmacytoid cells, often showing moderate to severe cytologic atypia, vesicular nuclei, and prominent nucleoli. High-grade morphology, characterized by severe nuclear atypia and increased mitosis, is indicative of poor prognosis. Immunoprofiling for these lesions involves the variable expression of cytokeratins, Epithelial Membrane Antigen (EMA), S100 protein, and often muscle/myoepithelial markers such as Smooth Muscle Actin (SMA) or calponin. Molecular studies have also demonstrated rearrangement in EWSR1 and SMARCB1 genes [7-9].
In this study, we present two cases of myoepithelial carcinoma. The first case involves a 46-year-old female with a nodule in the right hip region, and the second case involves a 58-year-old female with a swelling in the right thigh region.
Case 1
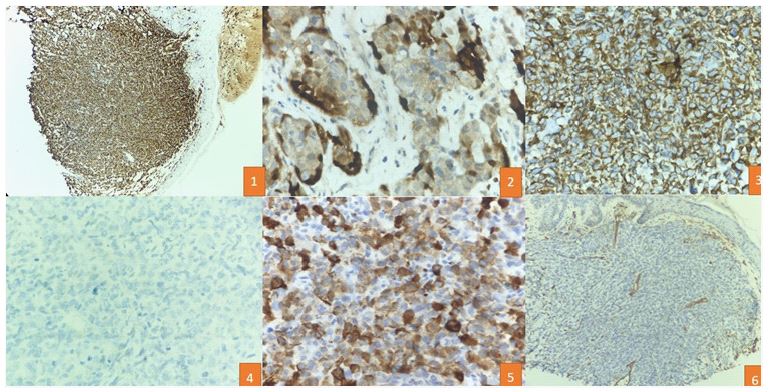
A 46-year-old woman developed a nodule on the right hip region that had grown rapidly in size over 2 years. Physical examination revealed a firm, non-tender, red nodular lesion with overlying skin pigmentation, measuring 5 x 4 cm. A palpable lymph node was found in the right inguinal region. Computed Tomography (CT scan) showed the tumor located in the dermis and subcutaneous tissue just above the muscle, while Positron Emission Tomography and Computed Tomography (PET scan) indicated uptake in the lesion and the right inguinal lymph node, with no distant metastasis. An initial excision biopsy was performed, revealing varied histological growth patterns, including solid sheet-like, tubulo-reticular, and myxoid stroma in the background, with zones of tumor necrosis. On higher magnification, the tumor cells displayed pleomorphism, with epithelioid, spindle, and plasmacytoid cells separated by thin-walled blood vessels. Ductal structures were also identified amidst the pleomorphic cells. The tumor cells exhibited vesicular to coarse chromatin, distinct nuclei, and brisk mitoses. No evidence of keratinization or melanin pigment was observed. Immunohistochemistry revealed diffuse strong p63 and S100 staining in the tumor cells, with positive pan-cytokeratin staining in pleomorphic cells. HMB45 and CD34 were negative in the neoplastic cells. The final impression suggested a carcinoma of myoepithelial origin with a focal element of epithelial-myoepithelial patterning. The patient underwent wide local excision and inguinal lymph node dissection with negative margins.
Case 2
A 58-year-old woman developed a swelling on the right thigh for 4 years. Physical examination revealed a 3 x 3 cm, non-tender, firm, red nodular lesion with pigmented skin overlay. No regional lymphadenopathy was observed. CT scan showed the tumor was located in the deep dermis and subcutis just above the muscle. An excision biopsy revealed various histological patterns, predominantly a tubule-reticular pattern with a myxoid stroma. On higher magnification, the tumor cells displayed pleomorphic spindle and plasmacytoid cells with vesicular to coarse chromatin, distinct nucleoli, and brisk mitoses. No evidence of keratinization or melanin pigment was observed. Immunohistochemistry showed diffuse and strong S100 positivity in the pleomorphic cells, with focal CK positivity suggestive of myoepithelial differentiation, while p63 was negative in these cells.
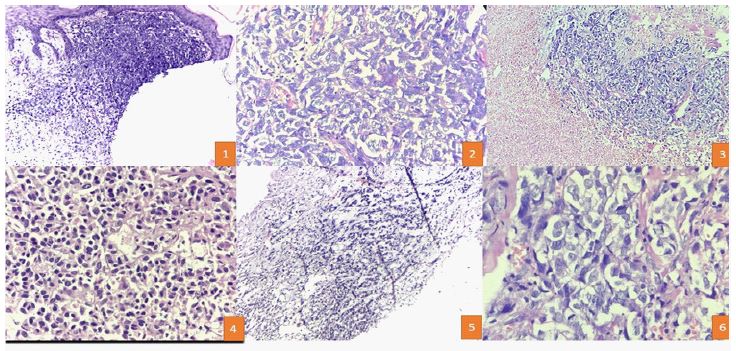
Discussion
Primary myoepithelial tumors of soft tissue, including mixed tumors, myoepitheliomas, and myoepithelial carcinomas, are rare and primarily occur in subcutaneous tissue. Limited demographic data on these tumors shows equal incidence in both sexes, with higher occurrence between the third and fifth dePrimary myoepithelial tumors of soft tissue, including mixed tumors, myoepitheliomas, and myoepithelial carcinomas, are rare and primarily occur in subcutaneous tissue. Limited demographic data on these tumors shows equal incidence in both sexes, with higher occurrence between the third and fifth decades of life [1]. Pediatric cases account for approximately 20% of occurrences, with the malignant counterpart being more common. While myoepitheliomas generally exhibit a benign course, with recurrence rates around 20% attributed to incomplete excisions or positive margins, myoepithelial carcinomas are more aggressive, with recurrence and metastasis occurring in 40-50% of cases [1].
This report highlights two cases of myoepithelial carcinoma occurring in the right hip and thigh regions, respectively. These tumors can arise in a wide range of anatomical locations, with extremities and proximal limb girdles being common sites. Like their counterparts in salivary glands, these tumors exhibit a spectrum of architectural and cellular diversity, showing various arrangements such as sheets, cords, reticular, and trabecular patterns, with epithelioid, spindled, clear, or plasmacytoid features, often surrounded by hyalinized or myxoid stroma [2]. Although previously referred to as Para chordoma, this terminology is not currently recommended in the WHO blue book.
The World Health Organization of Soft Tissue 2019 has established essential and desirable diagnostic criteria for myoepithelial carcinoma, including histological and immunohistochemical markers, along with genetic analysis in certain cases. Immunohistochemical patterns indicate reactivity to epithelial markers, with keratin AE/1-AE/3 or Pan-Keratin being the most sensitive epithelial markers (93% of cases), and calponin being the most sensitive among myogenic markers (86% of cases). Other markers, including smooth muscle actin, Glial Fibrillary Acidic Protein (GFAP), and p63, also contribute to accurate diagnosis [3].
In our cases, immunohistochemical analysis revealed positivity for S-100 and p63 and negativity for HMB45 and CD34. The varied patterns and location warranted excluding melanoma, while the abundance of small blood vessels necessitated ruling out a vascular tumor. The highlighted pan-cytokeratin in the ductal areas suggests a spectrum ranging from myoepithelioma to myoepithelial carcinoma. In challenging cases, the genetic profile of soft tissue myoepithelial tumors can be evaluated. EWSR1 gene rearrangements were seen in half the tumors outside the salivary glands, with partner fusion genes being POU5F1, PBX1, and ZNF444 [5-7].
The rarity of these lesions underscores the importance of a thorough assessment of morphology, along with an optimized immunohistochemical panel. In certain cases, confirmation of EWSR1 gene rearrangement may be necessary. The main modality of treatment is complete excision with clear margins, and adjuvant radiotherapy may be considered if tumor margins are positive [8]. Prognosis is generally favorable when surgical margins are free of tumor.
Conclusion
In conclusion, primary cutaneous myoepithelial lesions are rare entities with varying prognoses. Accurate diagnosis and proper management are crucial, supported by histology and an optimized immunohistochemical panel. Extensive research and literature review are necessary to explore therapeutic and prognostic targets in the context of personalized medicine. Given the limited number of Indian case reports on this entity, further studies are needed to expand our understanding of this rare condition in the Indian population [9-11].
References
- Hornick J, Fletcher C. Path myoepithelial tumors of soft tissue: A clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J SurgPathol. 2003; 27: 1183-96.
- Jo VY, Fletcher CD. Myoepithelial neoplasms of soft tissue: An updated review of the clinicopathologic, immunophenotypic, and genetic features. Head Neck Pathol. 2015; 9: 32-8.
- Soft Tissue and Bone Tumours WHO Classification of Tumours, 5th Edition, Volume 3 WHO Classification of Tumours Editorial Board. ISBN-13 978-92-832-4502-5.
- Rekhi B, Sable M, Jambhekar NA. Histopathological, immunohistochemical and molecular spectrum of myoepithelialtumours of soft tissues. Virchows Arch. 2012; 46: 687-97.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010; 49: 1114-24.
- Brandal P, Panagopoulos I, Bjerkehagen B, Heim ST (19; 22) (q13; q12). Translocation leading to the novel fusion gene EWSR1 ZNF444 in soft tissue myoepithelial carcinoma. Genes Chromosomes Cancer. 2009; 48: 1051 6.
- Brandal P, Panagopoulos I, Bjerkehagen B, Gorunova L, Skjeldal S, et al. Detection of at (1; 22)(q23; q12) translocation leading to an EWSR1 PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer. 2008; 47: 558 64.
- Neto AG, Pineda Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: A case report and review of the literature. Head Neck. 2004; 26: 470 3.
- Balraam KV, Shelly D, Mishra PS, Sharma I, Sampath KS, et al. Myoepithelial carcinoma of soft tissue: A report of two cases. Journal of Cancer Research and Practice. 2019; 6: 136.
- Tayal S, Suri V, Misra MC, Ray R. Myoepithelial carcinoma of soft tissue: a case report. Indian journal of pathology & microbiology. 2007; 50: 761-3.
- Swain N, Kumar SV, Pathak J, Patel S. Soft tissue myoepithelial carcinoma of neck: A rare case report with review of literature. Journal of Oral and Maxillofacial Surgery, Medicine, and Pathology. 2014; 26: 580-4.