
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
A case of Von Hippel-Lindau disease with multi-organ involvement
*Corresponding Author : Wondemagegnhu Tigeneh
Clinical Oncologist & Palliative Care Physician, Tikur Anbessa Specialized Hospital, P.O. Box 3819, Ethiopia. Tel: +251911897356
Email: Wondemagegnhu.tigeneh@aau.edu.et
tigeneh@yahoo.com
Received : Aug 26, 2023
Accepted : Sep 13, 2023
Published : Sep 20, 2023
Archived : www.jcimcr.org
Copyright : © Tigeneh W (2023).
Abstract
Von Hippel-Lindau (VHL) is an autosomal dominant disease with multi-organ involvement in familial neoplasm. Genetic aberrations of the tumor suppressor gene VHL cause it. Based on the reports in VHL, the most common initial presentation tumors are hemangioblastoma in the CNS and retina. In addition, patients can be present with signs and symptoms of other visceral organ involvement, such as the CNS, adrenal gland, the kidney, pancreas tumor and others.
Here we report a 34 years old woman presented with signs and symptoms of ICSOL secondary to hemangioblastoma, confirmed after the patient’s surgery. After years of the smooth postoperative period, she developed a carcinoid tumor at the head of the pancreas and left supra renal mass, for which she was operated and improved. Subsequent follow up detected recurrence and proper whippel surgery was done and confirmed the diagnosis of VHL.
From this case report easily can observe, regular and proper follow up lead early recognition and on-time appropriate treatment remain the mainstay of management in this group of patients.
Keywords: Von Hippel Lindau (VHL) disease; Hemangioblastoma; Multiorgan involvement.
Citation: Tigeneh W. A case of Von Hippel-Lindau Disease with Multi-Organ Involvement. J Clin Images Med Case Rep. 2023; 4(9): 2604.
Introduction
Von Hippel-Lindau (VHL) syndrome is an autosomal dominant, multi-organ, familial neoplastic syndrome caused by genetic aberrations of the tumor suppressor gene VHL located on chromosome [1-3]. The prevalence of VHL is estimated to be between 1:35,000 to 1:40,000. The mutation of the VHL gene leads to develop benign and malignant tumors of several visceral organs like the central nervous system, kidneys, pancreas, adrenal, and reproductive organs. The most common VHL-associated tumors are hemangioblastomas involving the brain, spinal cord, and retina; clear cell renal cell carcinoma (RCC); pheochromocytomas and paragangliomas of the adrenal gland and pancreatic neuroendocrine tumor. The current case report is a young woman with VHL disease involving hemangioblastoma of the posterior fossa followed by visceral multi-organ involvement.
Case report
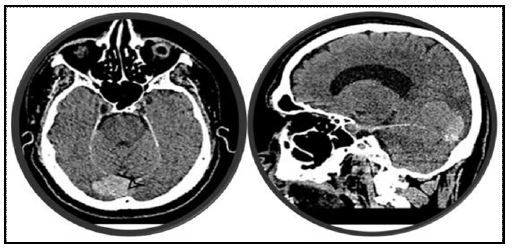
Here I am presenting a 34-year-old woman, a mother of two presented with a history of progressive global headache followed by nausea and vomiting of months duration. The intensity of the headache has increased, and it started to have an imbalance, followed by blurring of vision on and off, vertigo and ataxia of one week duration. She gave a history of long-standing dyspepsia and abdominal discomfort. After having an MRI of the brain, hemangioblastoma in the posterior fossa of the brain was diagnosed, and surgery was performed.
After eleven months of a smooth post-operative period, she started to experience dyspepsia, abdominal discomfort on the right side, anorexia, and weight loss. After investigation, a CT scan of the abdomen revealed a pancreatic head and suprarenal mass with multiple intra- abdominal lymphadenopathies with the impression of a carcinoid tumor and poorly controlled hypertension. She has been referred abroad (India), where the whipple procedure was done. Histopathology confirmed pancreatic neuroendocrine neoplasm PT2N1; all margins are negative one out of 12 LN +ve. She had regular follow-ups for almost two years, her blood pressure was well controlled, and after a while, she started to have anorexia, abdominal discomfort, itching sensation all over the body, and right-side abdominal swelling. On physical examination, poorly controlled hypertension (160/112 mmHg) and right upper quadrant abdominal tenderness were observed. Whole body PET-CT images were obtained with few somewhat defined enhancing lesions in the right suprarenal-retrocaval region showing mild tracer uptake; a tiny sub centimetric enhancing lesion involving the medial limb of the left adrenal gland the possibility of multiple pheochromocytomas is considered. Clinically relevant laboratory tests revealed as follows: Urine metanephrine/creatinine-141 μg/g Cret (Nr.29-158), normetanephrine/creatinine-22526 μg/g Crea (Nr.53-6590), serum serotonin (HPLC) 796++ ng/ml (Nr 68- 232), 5-HIAA (5-Hydroxy Indole AceticAcid), 24 hrs Urine 4.94 mg/24 hrs (WNL), Chromogranin A 185.5 ng/ml (Nr< 98.1), urine Epinephrine 2.57 μg/day (Nr.1.7-22.4), Norepinephrine 647.39 μg/day (Nr.12=1-85.5), and Dopamine 211.56 μg/day (Nr.52.00-480.00). Considering the recurrence of the carcinoid tumor, she was reoperated with adrenalectomy, lymphadenectomy, and omentectomy was done. The pathology result and Immunohistochemistry show’s morphological diagnosis of Neuroendocrine neoplasm, Synaptophysin all the tumor cells show cytoplasmic positivity, Chromogranin most of the cells show positivity, MIB-1-less than 1% of tumor cells and all consistent with neuroendocrine neoplasm grade I with proliferation index less than 1%.
Keywords: PCC: Pheochromocytoma; CNS: Central Nerve System; HB: Hemangioblastoma; RCC: Renal Cell Carcinoma
She had a smooth post-operative period without any complications; currently, almost five years after the patient’s last surgery, the blood pressure, as well as all symptoms, are well controlled, and she has been on regular follow up for the last five years without any sign of recurrence or progressive disease.
Discussion
Von Hippel-Lindau is a rare disease that is inherited in an autosomal dominant manner and causes multi-system involvement in the form of the development of hemangioblastoma of the Central Nervous System (CNS), retinal hemangioblastoma, renal cell carcinoma or renal cyst, endolymphatic sac tumor, neuroendocrine tumors and cyst of pancreatic gland, pheochromocytoma, epididymal cyst adenoma.
Hemangioblastoma of the CNS usually develops from childhood at any age of 10 or early teen until 30. These are benign tumors. Some hemangioblastomas remain unchanged in size over several years, and if they do not cause symptoms, their surgical removal may not be necessary. The symptoms of hemangioblastoma are usually caused by the expansion of tumors in the intracranial space and spinal cord. Asymptomatic small tumors are carefully watched until the onset of symptoms. Surgical resection is the best treatment modality for this tumor; in cases of enormous tumor burden where surgical resection is not possible, gamma knife surgery can substitute the treatment modality. The most common sites for hemangioblastoma development are the cerebellum and spinal cord, as evident in our patient. Our patient presented with signs and symptoms of cerebellar hemangioblastoma (CHb) and was operated on on time. Pathological confirmation was done, and routine investigations like organ function tests, abdomen ultrasound, and chest x-ray showed no abnormality. Unusual sites of hemangioblastomas in VHL include the anterior lobe of the pituitary, pituitary stalk, hypothalamus, optic nerve, corpus callosum, wall of the third ventricle, the temporal horn of the lateral ventricles, frontal and temporal lobe, meninges.
In VHL, Renal cysts are much more common than Renal Cell Carcinoma (RCC), with an incidence of 60% and 24-45% subsequently. Renal involvement in VHL is multi-centric and bilateral in at least 75% of patients microscopic solid tumors identified within the renal parenchyma of patients with VHL, which progress into giant macroscopic tumors. The renal tumor observed, on average, grow at the rate of 1.6 cm/year, which is somewhat faster than those in sporadic renal cell carcinoma [4-6].
In the VHL pancreas, different types of lesions like cysts, serious micro-cystic adenomas, and even malignant pancreatic lesions (adenocarcinoma) can be involved. The pancreatic cyst is the most common among the above lesions. Adrenal gland involvement in VHL having pheochromocytomas occurs only 7-18%. There are multiple case reports and involvement of different organs with VHL lesions like malignant lesions on the thyroid (Medullary and papillary type), liver and pulmonary hemangioblastoma, ovarian and omental cysts, hemangioma of skin, and skeletal muscles [7].
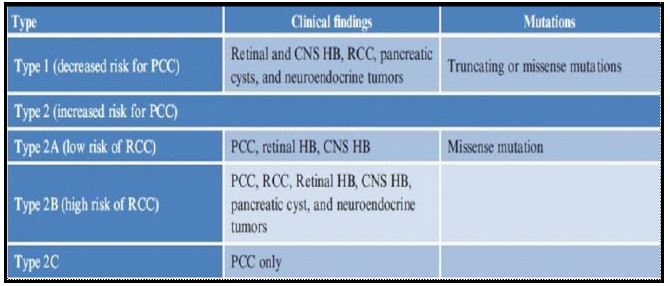
Patients can have variable presentations depending on the primary site; the diagnosis is often confirmed by positive family history and the presence of one VHL-associated tumor. In cases with no known history of VHL, multiple tumors are needed for diagnosis since approximately 20% of cases result from de novo mutations. Specific genotype–phenotype correlations in affected families led to the classification of VHL subtypes into types 1 and 2 (Table 1), primarily based on the presence of a pheochromocytoma. Type 1 disease has a shallow risk of pheochromocytomas. Type 2 VHL is further categorized into type 2a (low risk of RCC), type 2b (high risk of RCC), and type 2c, which only presents with pheochromocytomas.
Based on the above classification, our patient belongs to type 1 because she had CNS HB, pancreatic cystic mass, and neuroendocrine tumor. The sex and genotype of the patient influence survival. Female patients have a significantly higher risk of VHL-related death than male patients (HR=2.25, 95% CI 1.20 to 4.20, p=0.011). Overall, 79% (53 of 67) of deaths were VHL-related, but the risk of VHL-related death has decreased over time, as has the frequency of renal cell carcinoma (RCC)-related death. Surveillance is especially beneficial for truncating mutation carriers with the greatest RCC and Central Nervous System (CNS) hemangioblastoma risk.
Conclusion
The current study reinforces the importance of regular follow-ups with proper evaluation by appropriate diagnostic modalities. As we observed from our patient, these tumors often have multiple periods of tumors growth separated by periods of arrested growth, and different kinds of literature informed us the possibility of many untreated tumors might remain static for several years, yet follow-up of VHL patients must not be discontinued after their initial diagnosis and treatment. Advisable to make maximum effort to analyze the chromosomal mutation of the patient, which will help and lead us to offer the option of pre-symptomatic gene testing and early treatment. The advantage of regular follow-ups and checkups lead to early detection, which emphasizes better treatment outcomes.
References
- Keutgen XM, Hammel P, Choyke PL, Libutti SK, Jonasch E, et al. Evaluation and management of pancreatic lesions in patients with von Hippel-Lindau disease. Nature Rev Clin Oncol. 2016; 13: 537-49.
- Neumann HPH, Wiestler OD. Clustering of features of von Hippel Lindau syndrome: Evidence for a complex genetic locus. Lancet. 1991; 337: 1052-4.
- Maher ER, Iselius L, Yates JR, Littler M, Benjamin C, et al. Von Hippel Lindau disease: A genetic study. J Med Genet. 1991; 28: 443-447.
- Maher ER, Yates JRW, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA. Temporal Sequence. QJ Med. 1990; 77: 51-63.
- Quadery FA, Okamoto K. Diffusion-weighted MRI of haemangioblastomas and othercerebellar tumours. Neuroradiology. 2003; 45: 212-219.
- Choyke PL, Glenn GM, Walther MM, Zbar B, Weiss GH, et al. The natural history of renal lesions in Von HippelLindau disease: a serial CT imaging study in 28 patients. AJR. 1992; 159: 1229-1234.
- Melmon KL, Rosen SW. Lindau’s disease. Am J Med. 1964; 36: 595-617.
- Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, et al. Genotype-phenotype correlations in von Hippel‐ Lindau disease. Hum Mutat. 2007; 28: 143-9.
- O’ Brien FJ, Danapal M, Jairam S, Lalani AK, Cunningham J, et al. Manifestations of Von Hippel Lindau syndrome: A retrospective national review. QJM. 2014; 107: 291-6.
- Binderup ML, Jensen AM, Budtz-Jørgensen E, Bisgaard ML. Survival and causes of death in patients with von Hippel-Lindau disease J Med Genet. 2017; 54: 11-18.