
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Granulomatosis with polyangiitis: The wily masquerader of cavitary pulmonary lesions
Sona Mitra1*; Ravish Kshatriya2; Ashish Bavishi3; Suman Shil4; Jeet Patel5; Kanugir Gosai6; Amal Bhattacharya7
1Consultant Physician, Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
2Professor and HOD, Department of Respiratory Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
3Infectious Diseases Specialist, Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
4Assistant Professor, Department of Respiratory Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
5Rheumatologist, Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
6Professor, Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
7Professor and HOD, Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
*Corresponding Author : Sona Mitra
Department of Medicine, PIMSR, Parul University, Waghodia, Vadodara, Gujarat, India.
Email: sonamitra22@gmail.com
Received : Sep 15, 2023
Accepted : Oct 09, 2023
Published : Oct 16, 2023
Archived : www.jcimcr.org
Copyright : © Mitra S (2023).
Abstract
Background: Granulomatosis with Polyangiitis (GPA) is a small and medium vessel vasculitis with predominant respiratory and renal involvement that causes florid presentation of the disease entity with varying spectrum of illness at different time intervals.
Case: We report the case of a 42-years-old female patient with symptoms of low-grade fever, productive cough and significant loss of weight with multiple cavitary lesions on Chest radiograph. The patient was initially suspected to have tuberculosis which was eventually ruled out. A diagnosis of vasculitis was suspected as patient gave history of repeated hospitalisations with symptoms of upper respiratory tract involvement, left facial nerve palsy and sensorineural hearing loss, A diagnosis of limited GPA was made when the cytoplasmic Antineutrophil Cytoplasmic Antibodies, (c-ANCA) and antiproteinase-3 antibodies (anti-PR3) was elevated. The patient was treated with combination immunosuppressive therapy of prednisolone and methotrexate which induced clinical remission within one month and complete radiological remission within 4 months.
Conclusion: GPA is an important non infective cause of cavitary pulmonary lesions that presents with extrapulmonary clinical spectrum (recurrent sinusitis, ear infections, renal dysfunction and neurological disease). Multifocal presentation of the disease at different time intervals results in considerable delay in diagnosis and adds to the morbidity of the disease entity.
Keywords: Granulomatosis with polyangiitis; Vasculitis; Cavitary lesions; Immunosuppressive therapy.
Abbreviations: GPA: Granulomatous Polyangiitis; c-ANCA: Cytoplasmic Antineutrophil Cytoplasmic Antibodies; anti PR3: Antiproteinase-3 antibodies; anti MPO: Myeloperoxidase Antibodies.
Citation: Mitra S, Kshatriya R, Bavishi A, Shil S, Patel J, et al. Granulomatosis with polyangiitis: The wily masquerader of cavitary pulmonary lesions. J Clin Images Med Case Rep. 2023; 4(10): 2643.
Introduction
Granulomatosis with polyangiitis, formerly known as Wegener’s Granulomatosis, is a rare small to medium vessel multisystem necrotizing vasculitis which typically involves respiratory and renal system [1,2]. It was first described by German pathologist Friedrich Wegener in 1936. American College of Rheumatology requires 2 of 4 criteria for diagnosis: Positive biopsy for granulomatous vasculitis, urinary sediment with red blood cells, abnormal chest radiograph and oral/nasal inflammation. It has no gender preponderance and affects patients between ages of 40 to 55-years in more than two thirds of cases [3]. The aetiology of the disease remains unclear but a strong association with Antineutrophil Cytoplasmic Antibodies (ANCAs) in immunofluorescence assays directed against proteinase-3 and myeloperoxidase has been found [4,5]. Here, we present a case of Granulomatosis with polyangiitis presenting with upper respiratory tract, neurological and pulmonary manifestations.
Case report
Our patient, a 43-year-old woman with no pre-existing comorbidities presented with complaints of low grade fever, headache, chronic productive cough, loss of appetite and involuntary weight loss of more than 7 kg over 3 months period. In the past 8 months, she had multiple ENT consultations for nasal discharge and chronic headache. Non contrast CT scan of head and neck showed pansinusitis for which she had undergone Functional Endoscopic Sinus Surgery (FESS). 2 months later she developed left sided otalgia and otorrhea which did not respond to antibiotic therapy. CT Temporal bone was done which was suggestive of chronic oto-mastoiditis. She was operated for the same with left cortical mastoidectomy and type II tympanoplasty. Tissue obtained from antrum revealed fibro collagenous tissue with scattered lymphocytes, focal necrosis with hyalinosis. She presented to a neurologist with left facial paralysis after surgery. She also presented with 6 months history of progressive hearing loss and was diagnosed with bilateral sensorineural deafness. MRI Brain with MRA was normal.
Further detailed history revealed that our patient was a non-smoker; there was no past history of Tuberculosis, occupational or environmental exposures to explain a chronic respiratory illness.
Physical examination findings were relatively unremarkable. Pulse rate was 100/min, blood pressure was 130/70 mmHg, respiratory rate was 16/min, body temperature was 99.4°F and oxygen saturation (SO2) level was 98% on room air, as measured by pulse oxymetry. There was no skin rash, nose deformity, oral ulcers, joint tenderness or swelling. Systemic examination was unremarkable.
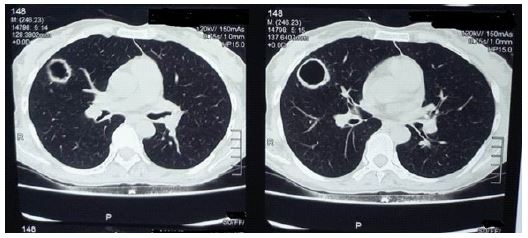
Routine and specific laboratory test results findings were as follows. The patient had microcytic hypochromic anaemia (Hemoglobin -10.2 g/dl), thrombocytosis (Platelet count -4.82×105/μL, leukocytosis (WBC count- 18.9×103 cells/μL, neutrophil predominant-72%), elevated ESR (102 mm/hr) and CRP (149.60 mg/L). Chest X-Ray showed B/L thick walled cavitary lesions in upper and middle lobes (Figure 1). HRCT Thorax revealed bronchiectatic cavitary lesions in B/L upper and middle lobes (Figure 2). There was no accompanying mediastinal lymphadenopathy or pleural effusion seen. Sputum for acid fast bacilli (AFB), gram stain, KOH mount and culture was negative. An Infectious Diseases opinion was sought to rule out other causes of infective etiology. Bronchoscopy with BAL was advised along with ANA, p-ANCA and c-ANCA. BAL fluid was sent for AFB, culture sensitivity, CB-NAAT, KOH mount, Fungal culture, MGIT TB culture, and Galactomannan Index (GMI) which were inconclusive. Nasal biopsy showed nonspecific chronic inflammatory cells.
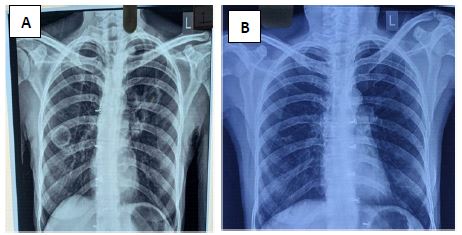
Simultaneously, the patient was found to have a positive c-ANCA with a high titre of anti-Proteinase-3 (anti-PR3) at 5.3 U/ml (normal range < 1.0 U/ml) and a negative anti Myeloperoxidase (anti-MPO). She was diagnosed with GPA with pulmonary involvement preceded by the upper respiratory tract disease (sinusitis and ear involvement) and facial nerve palsy. She was counselled regarding the disease course and treatment and was subsequently treated with prednisolone 60 mg daily (1 mg/kg) and oral methotrexate 10 mg per week with folinic acid supplementation. Within the first few weeks of treatment, there was considerable resolution of respiratory symptoms, appetite improved and weight gain was observed. The patient was followed up fortnightly and the dose of oral methotrexate was gradually increased to a maximum dose of 20 mg per week after 8 weeks. Prednisolone was also gradually stepped down to (0.5 mg/kg), 30 mg daily within 12 weeks. There were no adverse drug reactions noted. At the end of 16 weeks, the patient remained asymptomatic with a remarkable weight gain to the baseline state. Sensorineural deafness persisted post 16 weeks of therapy. Repeat Chest X-ray at the end of 4 months showed complete resolution of all lung cavities ( Figure 1B). After multidisciplinary consensus, it was agreed to continue prednisolone at 10 mg daily and methotrexate at 20 mg per week. The improvement in pulmonary symptoms was sustained on subsequent follow up and the disease has been in remission for a period of 12 months.
Discussion
Granulomatosis with polyangiitis, is a multi-system necrotizing non-caseating granulomatous vasculitis affecting small and medium arteries, capillaries and veins. Clinically, the patients may complain of a wide spectrum of manifestations. Upper respiratory tract manifestations are common and includes nasal obstruction, rhinitis, epistaxis, sinusitis, mastoiditis and otitis media [6,7]. Sino-nasal mucosal ulcers, nasal septal perforation, nasal saddle deformity or subglottic stenosis have also been described in the literature [7].
Pulmonary involvement may be seen in upto 85% cases. Radiographic presentation of pulmonary manifestations is categorized into four patterns [7,8]. Most common radiologic presentation includes randomly distributed multiple nodules of different sizes throughout the lungs with or without central cavitation [7,8]. Second most common pattern includes multifocal regions of consolidation or ground glass opacities reflecting pulmonary haemorrhage [7,8]. Reticulonodular pattern, peripheral wedge-like consolidation and pleural effusion have also been described in the literature [7].
Approximately 50% of patients may have renal involvement at clinical presentation, which are characterized by reduced renal function, proteinuria and hematuria [6]. Central Nervous System (CNS) manifestations are reported in approximately 5% of the patients which include hypertrophic pachymeningitis, small vessel vasculitis resulting in infarcts or arterial occlusion, and intracranial haemorrhage [1,7]. Ophthalmic manifestations have been reported in 40%-50% of patients [1]. Small vessel vasculitis component may result in conjunctivitis, scleritis, episcleritis, uveitis, optic neuritis, optic nerve vasculitis or retinitis.It can also result in an orbital granulomatous non-caseating inflammatory mass resulting in proptosis with or without optic nerve compression [1,7].
In our case report, we describe a lady in her early 40’s with low grade fever, headache, chronic productive cough, loss of apetite and loss of weight with radiological appearance of multiple cavitary lesions. She gave history of repeated admissions in the past for symptoms of upper respiratory tract involvement. The repeated upper airway involvement (sinusitis, mastoiditis and hearing impairment) that was seen in our patient could have been an important clue to clinching the diagnosis of GPA much earlier. It has been reported that upper airway involvement can be present in patients with GPA months before manifestation of pulmonary or renal disease [9]. Most patients report mild non-specific constitutional symptoms before developing severe disease (cavitary pneumonia and/or acute renal failure). Therefore, the diagnosis of GPA is a race against time before a full-blown spectrum of illness precipitates, which results in not only considerable diagnostic challenge, but also disease related morbidity and chronic sequelae.
Laboratory tests show elevated levels of Anti-Neutrophil Cytoplasmic Antibodies (ANCA) with a cytoplasmic staining pattern directed against Proteinase 3 (PR3). Being related to disease activity, ANCAs have been identified as risk factors of GPA relapse and are currently used in the long-term follow-up process [10]. Histologically, evidence of necrotizing granulomas usually indicates the diagnosis; however, treatment can be initiated even if a histological diagnosis cannot be made, if the clinical criteria of diagnosis are present and the c-ANCA titre is positive.
The current standard of care for GPA includes high dose corticosteroids combined with cyclophosphamide, methotrexate or rituximab. In majority of cases, it is suggested that the treatment should be continued for a minimal period of 18 months but shorter treatment regimens have also been suggested to minimize risks of infection [5,11]. Majority of patients achieve remission within 3-6 months with conventional treatment, but nearly half of them relapse when treatment is de-escalated or stopped [3,5]. In cases with high risk of relapse, treatment should be extended for up to 24 months [1]. Although, c-ANCA/anti PR3 titres is being used greatly to predict the risk of relapse in such patients, over-reliance on this parameter is greatly disputed [12].
Although we could not make a histopathological diagnosis of pulmonary lesions (i/v/o thin, friable cavitary wall) in our patient, the clinical history showing involvement of upper respiratory tract, followed by involvement of lungs with raised c-ANCA levels, made the likelihood of diagnosis of GPA high which was confirmed with good response to immunosuppressive therapy. Although she has been in remission for a period of 12 months, we plan to continue low dose prednisolone and maintenance methotrexate at least 18-24 months.
Conclusion
GPA, an autoimmune granulomatous small and medium vessel vasculitis, is an important non infective cause of cavitary pulmonary lesions that presents with extrapulmonary clinical spectrum (recurrent sinusitis, ear infections, renal dysfunction and neurological disease). Multifocal presentation of the disease at different time intervals results in considerable delay in diagnosis and adds to the morbidity of the disease entity.
References
- Weiner M, Segelmark M. The clinical presentation and therapy of diseases related to anti-neutrophil cytoplasmic antibodies (ANCA). Autoimmun Rev. 2016; 15: 978-82.
- Ramponi G, Folci M, De Santis M, Damoiseaux JGMC, Selmi C, et al. The biology, pathogenetic role, clinical implications, and open issues of serum anti-neutrophil cytoplasmic antibodies. Autoimmun Rev 2021; 20: 102759.
- McCabe C, Jones Q, Nikolopoulou A, Wathen C, Luqmani R. Pulmonary-renal syndromes: An update for respiratory physicians. Resp Med. 2011; 105: 1413-1421.
- Langford C. Clinical features and diagnosis of small-vessel vasculitis. Cleve Clin J Med. 2012; 79: S3-7.
- Anthony AIL, Ibrahim ZA, Li-Cher L. From Triumph to Tribulation: A Granulomatous. Polyangiitis Case Report. Int J Respir Pulm Med. 2019; 6: 113.
- Greco A, Marinelli C, Fusconi M, Macri GF, Gallo A, et al. Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopathol Pharmacol. 2016; 29: 151-9.
- Lakhani DA, Balar AB, Adelanwa A, Gross A, Mohamed R, et al. Granulomatosis with polyangiitis: A case report and brief review of literature. Radiol Case Rep. 2021; 16: 3445-3450.
- Feragalli B, Mantini C, Sperandeo M, Galluzzo M, Belcaro G, et al. The lung in systemic vasculitis: radiological patterns and differential diagnosis. Br J Radiol. 2016; 89: 20150992.
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: Prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983; 98: 76-85.
- Trandafir CM, Balica NC, Horhat DI, Mot IC, Sarau CA, et al. Granulomatosis with Polyangiitis (GPA)-A Multidisciplinary Approach of a Case Report. Medicina (Kaunas). 2022; 58: 1837.
- McGregor JG, Hogan SL, Hu Y, Jennette CE, Falk RJ, et al. Glucocorticoids and relapse and infection rates in anti-neutrophil cytoplasmic antibody disease. Clin J Am Soc Nephrol. 2012; 7: 240-7.
- Specks U. Controversies in ANCA testing. Cleve Clin J Med. 2012; 79: S7-11.