
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Hematopoietic stem cell transplantation in primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: A case report
Lucía Sopena*; Miriam Merchante; Maria Aranzazu Alcácera; Isabel Varela; Raquel Fresquet; Aritz Merchán; Maria Reyes García; Elia Chilet; Beatriz Bonaga; Tránsito Salvador
Lozano Blesa University Clinical Hospital, Zaragoza, Spain.
*Corresponding Author : Lucía Sopena
Pharmacy Department, Lozano Blesa University Clinical Hospital, San Juan Bosco Avenue, 15. 50009, Zaragoza, Spain.
Tel: +34 608433036;
Email: luciasopenacarrera@gmail.com
Received : Sep 18, 2023
Accepted : Oct 10, 2023
Published : Oct 17, 2023
Archived : www.jcimcr.org
Copyright : © Sopena L (2023).
Abstract
Primary Cutaneous Aggressive Epidermotropic CD8+ T-Cell Lymphoma (PCAETCL), a rare subtype of Primary Cutaneous Lymphoma (PCL), is known for its aggressive clinical course and refractoriness to standard treatments. Clinically, PCAETCL often presents with skin lesions that rapidly disseminate to various extracutaneous sites, resulting in a grim prognosis with a 5-year survival rate of less than 40%.
This article presents a case of a 43-year-old male patient initially diagnosed with CD8+ cytotoxic Mycosis Fungoides (MF). The patient’s treatment course involved multiple therapeutic modalities, including methotrexate, bexarotene, gemcitabine, peginterferon alfa 2a, and radiation therapy. Despite several lines of treatment, disease progression persisted. Eventually, the patient underwent allogeneic Hematopoietic Stem Cell Transplantation (allo-HSCT) from his sister, marking a turning point in his therapeutic approach.
The case underscores the diagnostic and therapeutic challenges associated with PCAETCL. It shows the heterogeneous responses found in literature, ranging from brief remissions to unfavorable outcomes, including severe infections. This highlights the lack of standardized treatment protocols for PCAETCL, with HSCT, both allogeneic and autologous, emerging as potential options for achieving prolonged responses.
In conclusion, PCAETCL remains a rare and aggressive clinical entity with no established standard treatment. This case report illustrates the aggressive nature of the disease and the importance of individualized treatment approaches, including transplantation. Collaborative research efforts are crucial to enhance our understanding of PCAETCL, identify prognostic factors, and develop more effective therapeutic strategies to improve patient outcomes.
Keywords: Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma Lymphoma; T-Cell; Cutaneous; Allogeneic hematopoietic stem cell transplantation Mycosis Fungoides; Extracutaneous manifestation Central nervous system involvement Multiple lines of therapy.
Abbreviations: PCAETCL: Primary Cutaneous Aggressive Epidermotropic CD8+ T-Cell Lymphoma; PCL: Primary Cutaneous Lymphoma; PET-TC: Positron Emission Tomography and Computerised Tomography; PUVA: Psoralen-Ultraviolet Radiation of Long Wavelength.
Citation: Sopena L, Merchante M, Alcácera MA, Varela I, Fresquet R, et al. Hematopoietic stem cell transplantation in primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: A case report. J Clin Images Med Case Rep. 2023; 4(10): 2646.
Introduction/background
Primary Cutaneous Lymphoma (PCL) is a type of non-Hodgkin lymphoma with an incidence of 1 per 100,000 worldwide. T-Cell PCLs (CTCLs) account for 75-80% of all PCL. Mycosis Fungoides (MF) is the most frequent subtype (60%) of CTCLs [1].
MF is considered an epidermotropic CD4 T-cell lymphoma, although it can also occur in cytotoxic TCD8s. This kind of MF is characterised by an indolent course of slow progression over years in the form of skin lesions such as patches, plaques and tumours, and in more advanced stages may develop lymph node and/or visceral progression [2]. At the present, there is no consensus on treatment. Pharmacoterapeutic options range from topical treatment (retinoids and phototherapy) to systemic treatment with mono- or polychemotherapy and targeted drugs [3,4]. Responses to treatment are usually transient, short-lived, and in advanced stages with successive relapses. Drug treatment aims to control the disease. At the present, the only curative treatment is allogeneic haematopoietic cell transplantation (allo-HSCT) [2-5].
Primary Cutaneous Aggressive Epidermotropic CD8+ T-Cell Lymphoma (PCAETCL) or Berti lymphoma is a rare (< 1%) subtype of CTCLs, described in 1999. It predominates in adult males and manifests as cutaneous lesions that within weeks or months show dissemination to other sites such as the lung, testes, adrenal glands, Central Nervous System (CNS) or oral cavity [2].
Histopathologically, it is characterised by the presence of an infiltrate of atypical medium-sized CD8+ lymphocytes, with epidermotropism in a pagetoid pattern. It has an aggressive behaviour and a poor prognosis, with a 5-year survival of less than 40%. Although this entity is a therapeutic challenge, initial treatment is based on polychemotherapy, but the only option with more prolonged responses is HSCT, which should be considered in young-fit patients since the moment of diagnosis.
We present the case of a male patient initially diagnosed with MF. Due to the occurrence of aggressive symptoms and dissemination to the CNS that emerged suddenly, was re-diagnosed as PCAETCL. In this case report we present the pharmacotherapeutic management carried out by a multidisciplinary team including numerous lines of therapy, relapses and adverse reactions. After receiving numerous unsuccessful lines of treatment, the patient was transplanted with allo-HSCT.
Case presentation
A 43-year-old male patient, with no medical history of interest, was diagnosed in October 2019 with CD8+ cytotoxic MF stage T3N0M0B0 (stage IIB). The PET-CT scan showed no lymphadenopathy with metabolic uptake. Treatment was started with subcutaneous (sc) methotrexate 12.5 milligrams (mg) per week, progressively increasing to 25 mg per week, associated with oral (po) bexarotene 225 mg every 21 days and topical corticosteroid treatment with clobetasol on the lesions. There was a significant improvement in the lesions, which reduced in size. The patient also reported good tolerance to the treatment. In March 2020, for better control of remaining lesions, PUVA was associated by two/three times a week receiving only one dose because of the impossibility due to SARS-coV-2 pandemic.
The disease stayed stable until March 2021. After that, ulcerated and very painful skin lesions appeared again, confirming progression with PET-CT in April 2021. It was decided to start rescue treatment with topical imiquimod 5%, which was discontinued due to the patient did not tolerated it. For this reason, it was decided to start with intravenous (iv) gemcitabine 1000 mg/m2 every 21 days associated with radiotherapy (RT) receiving 6 cycles (last July 2021) with good tumour response and good tolerance.
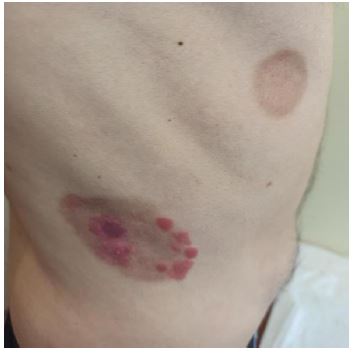
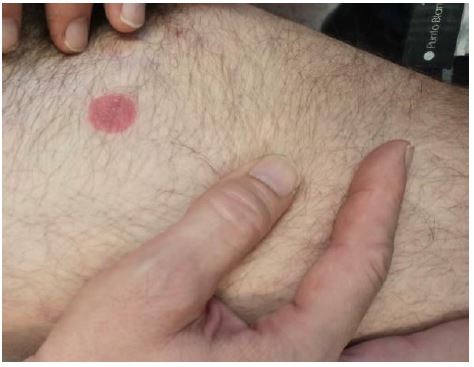
In August 2021, new lesions appeared, so it was decided to start treatment with sc peginterferon alfa 2a (off-label) 180 micrograms (mcg) per week, which was finally discontinued due to poor tolerance and poor response (Figures 1,2).
In November 2021, after new progression, he restarted iv gemcitabine with the aim of achieving complete remission prior to allo-HSCT, but in January 2022 progression appeared in the form of new fast-growing skin tumours. These lesions were biopsied and MF was found in tumour phase with aberrant expression of CD20 and negativity of CD30 with a high proliferative index. Sc methotrexate 25 mg per week was started again along with RT, ending in March 2022 with an apparent good response of the tumour lesions, although some patches and plaques persisted.
In June 2022 the patient was offered the possibility of participating in a clinical trial with tinotamustine. The patient accepted, but after several doses, he decided to leave the clinical trial due to the lack of efficacy and very poor tolerance to the drug.
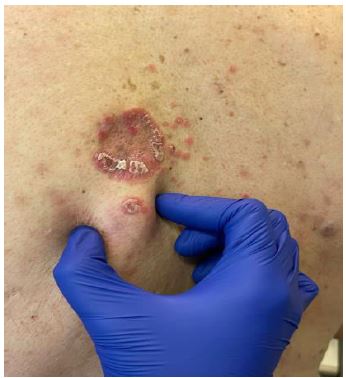
In October 2022, the lesions were rebiopsied and some of them were found to be positive for CD30, so it was decided to start treatment with iv Brentuximab Vedotin 1.8 mg/kg in monotherapy every 21 days. After 3 cycles of no response and the appearance of tumour lesions on the back (Figure 3), it was decided to reintroduce methotrexate and RT.
In December 2022, the patient was admitted to hospital suffering from covid-19 infection. At his hospital discharge, he began with bilateral hypoacusis, accompanied by vertiginous syndrome and later facial paralysis. For this reason, a lumbar puncture was performed and infiltration of the CNS was found. Given the observed aggressiveness of the tumour and the CNS infiltration, it was considered the possibility of being another type of more aggressive cutaneous lymphoma. A sample of the tumour was sent for genetic and molecular analysis. The result of the genetic study of the tumour showed the mutation PICALM(chr11,8 56 8 5751,-): JAK2(chr9,50 8 0531,+) (Score: 0,9 52).
The case was re-evaluated by the hospital’s Tumour Committee and due to the evolution of the clinical condition and the results of the molecular study of the tumour, PCAETCL lymphoma was re-diagnosed.
Thus, in December 2022 the patient started systemic chemotherapy treatment with CHOP (cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2, prednisone 60 mg/m2) with intrathecal therapy (ITT) (methotrexate 15 mg, cytarabine 40 mg and hydrocortisone 20 mg). As a result, there was an improvement of the left ear and vertigo, but poor improvement of facial paralysis.
In January 2023, he started a iv MATRIX regimen (rituximab 375 mg/m2, thiotepa 30 mg/m2, cytarabine 2000 mg/m2, methotrexate 3500 mg/m2). On 11 February the patient suffered methotrexate intoxication, reaching a level of 39.86 micromol/litre (μM) and a creatinine value of 2.48 mg/dL, which was managed with folinic acid and required the iv administration of the antidote glucarpidase 48 hours post-administration. The patient got back to normal methotrexate levels (< 0.01 μM) after 12 days. Levels of creatinine were recovered (< 1 mg/dL) at 15 days after methotrexate administration.
The administration of the treatment was delayed due to the need of waiting to get the patient a clinical stable situation. During this period, new erythematous skin lesions appeared, making the transplant an urgent option.
Two compatible siblings of the patient (HLA 9/10) were visited and assessed.
Once the patient got a clinical stable condition, he started transplant conditioning therapy with the TBF regimen (thiotepa 5 mg/kg, busulfan 2.7 mg/kg, fludarabine 30 mg/m2) and was transplanted with allo-HSCT from his sister on 24 March 2023.
40 days after transplant the patient had grade I cutaneous Graft-versus-Host Disease (GvHD) in the form of a micropapular rash and after 64 days acute gastrointestinal GvHD as diarrhea of 5 stools a day and persistent nausea and vomiting.
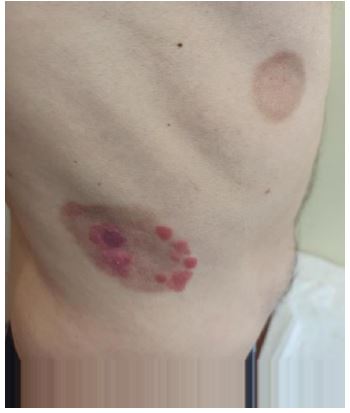
In June 2023, the patient suffered a relapse with the appearance of a new tumor lesion in the forearm (Figure 4). Oral ruxolitinib 10 mg every 12 hours per day was started.
The patient is still alive today (September 2023).
Discussion
PCAETCL is a rare and aggressive form of T-cell lymphoma that primarily affects the skin, characterised by infiltration of CD8+ cytotoxic T-lymphocytes in the epidermis. In some cases, PCAETCL may present with extracutaneous manifestations, such as CNS infiltration, which worsens prognosis and response to treatment. It represents a particularly challenging clinical entity due to its unfavourable prognosis and lack of effective treatment options.
The present report shows the case of patient with PCAETCL initially diagnosed as MF, who was subsequently re-evaluated due to the aggressiveness of the lymphoma and CNS infiltration. Multiple lines of therapy were employed, including corticosteroids, retinoids, methotrexate, targeted therapy, chemotherapy and intrathecal chemotherapy for CNS involvement. Subsequently, an allogeneic HSCT was performed from his sister, and the patient is still alive today.
Literature shows several cases of cytotoxic epidermotropic lymphomas with unfavourable prognosis and short survival were found. For example, a case reported in the Chilean population showed extreme aggressiveness from admission, refractoriness to treatment with corticosteroids, retinoids and first- and second-line chemotherapy, and very short survival rate [6]. Another case described a 69-year-old woman with PCAETCL and aberrant phenotype, who was treated with corticosteroids, bexarotene, methotrexate associated with radiotherapy, and one course of liposomal doxorubicin. At the end, she died of a Staphylococcus aureus skin infection leading to sepsis [7].
In addition, cases were found in which the initial diagnosis was erroneous, delaying appropriate treatment. For example, a case was reported of a young woman who was initially diagnosed with febrile ulceronecrotic Mucha-Habermann disease and treated with methylprednisolone and methotrexate. Subsequently the diagnosis was revised to PCAETCL because she developed respiratory symptoms. The patient died after 15 days [8].
The case we present is similar to that presented by Hajer Oun, who after receiving iv methotrexate and 6 cycles of CHOP together with radiotherapy, developed diplopia, aphasia and facial drooping, confirming infiltration of CD8+ lymphocytes in the CNS. The patient received intrathecal methotrexate and high doses of iv methotrexate and cytarabine [9].
In the available literature, cases of patients who have received HSCT are described, such as a case reported by Brüggen MC et al. who received 6 cycles of R-CHOEP with complete remission and subsequent allogeneic transplantation. The patient achieved a survival of 9 months, but then died of progression and sepsis [7]. Sarauta H et al. reported another example of a patient who received chemotherapy and TASPE but later died from acute myeloid leukaemia related to the treatment he had received [10].
In contrast to these unfavourable cases, some cases were identified in which a durable remission or therapeutic success was achieved after HSCT. For example, one patient with PCAETCL received intensive chemotherapy followed by allo-HSCT and 36 months later the patient is disease-free and in good functional status [11]. Another patient treated with aggressive chemotherapy followed by allo-HSCT and brentuximab vedotin after post-transplant relapse achieved a durable remission [12]. Another patient relapsed after autologous transplantation but then received allogeneic transplantation [13].
On the other hand, Al Aoun SM and their colleagues published in 2018 the first case of PCAETCL that achieved a durable remission of 58 months receiving only one line of treatment consisting of aggressive chemotherapy (hyper-CVAD cycle) [14].
As it was seen in the aforementioned case reports in the literature, there is no defined standard treatment for PCAETCL, but rather contrasting therapeutic options, and HSCT (allo or auto) is an alternative that may lead the patient to more prolonged responses.
Conclusion
PCAETCL is a rare and aggressive clinical entity that presents significant diagnostic and treatment challenges. Although cutaneous lymphomas represent a small proportion of all lymphomas, PCAETCL stands out for its aggressive behaviour and lack of effective therapeutic options.
The presented clinical case reflects the aggressive, systemic course and refractoriness to treatment commonly seen in patients with PCAETCL. Despite multiple prior lines of treatment resulting in therapeutic failure, the patient ultimately underwent a transplant, which is an example of successful management of this disease. However, the patient of this report suffered complications and relapsed 3 months post-transplant.
There are cases in the literature demonstrating positive responses to different therapeutic approaches, such as the use of intensive chemotherapy, radiotherapy and HSCT. Nevertheless, complications and unfavourable outcomes, such as relapse and death due to severe infections, have also been documented.
Given the small number of cases of PCAETCL, randomised clinical trials are limited. However, the collection and analysis of isolated clinical cases, such as the one presented here, can provide valuable information to guide the management of this pathology.
Further research and collaboration is needed to better understand the pathogenesis of this disease, identify prognostic factors, and develop more effective treatment strategies to improve outcomes in patients with PCAETCL.
Acknowledgements: The authors have no conflicts of interest to declare.
References
- Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood. The American Society of Hematology. 2019; 133: 1703-14.
- Calzado L, Fernández R, García A, López I, Marín A, et al. Guía Multidisciplinar para el abordaje diagnóstico y terapéutico del paciente con linfoma cutáneo primario. Madrid: Treelogy Medical Marketing S.L. 2021.
- Howles A, Scarisbrick J. Cutaneous T-cell lymphoma: Practical recommendations to enhance clinical practice. Br J Hosp Med (Lond). 2021; 82: 1-6.
- Coronado M, De la Cruz F, Gutiérrez AM, Martín A, Novelli S. Guía de GENTALMO para el diagnóstico y tratamiento de los linfomas T. Madrid: Treelogy Medical Marketing S.L. 2017.
- Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M; ESMO Guidelines Committee. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; 29: iv30-iv40.
- Intriago BM, Peña OC, Gray HAM, Cabrera CME, Martínez CV. Primary cutaneous aggressive epidermotropic cytotoxic CD8 positive T cell lymphoma: Report of one case. Rev Med Chil. 2012; 140: 368-72.
- Brüggen MC, Kerl K, Haralambieva E, Schanz U, Chang YT, et al. Aggressive Rare T-cell Lymphomas with Manifestation in the Skin: A Monocentric Cross-sectional Case Study. Acta Derm Venereol. 2018; 98: 835-841.
- Sheng N, Li Z, Su W, Liu W, Zong W, et al. A Case of Primary Cutaneous Aggressive Epidermotropic CD8+ Cytotoxic T-cell Lymphoma Misdiagnosed as Febrile Ulceronecrotic Mucha-Habermann Disease. Acta Derm Venereol. 2016; 96: 136-7.
- Oun H, Wilson M, Leach M, McKay P. Central nervous system relapse of primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Br J Haematol. 2021; 193: 9.
- Saruta H, Ohata C, Muto I, Imamura T, Oku E, et al. Hematopoietic stem cell transplantation in advanced cutaneous T-cell lymphoma. J Dermatol. 2017; 44: 1038-1042.
- Wehkamp U, Glaeser D, Oschlies I, Hilgendorf I, Klapper W, et al. Successful stem cell transplantation in a patient with primary cutaneous aggressive cytotoxic epidermotropic CD8+ T-cell lymphoma. Br J Dermatol. 2015; 173: 869-71.
- Cyrenne BM, Subtil A, Girardi M, Foss F. Primary cutaneous aggressive epidermotropic cytotoxic CD8+ T-cell lymphoma: Long-term remission after brentuximab vedotin. Int J Dermatol. 2017; 56: 1448-1450.
- Liu V, Cutler CS, Young AZ. Case records of the Massachusetts General Hospital. Case 38-2007. A 44-year-old woman with generalized, painful, ulcerated skin lesions. N Engl J Med. 2007; 357: 2496-505.
- Al Aoun SM, Iqbal S, AlHalouli TM, Zaidi SZ, Motabi IH. Durable remission of a patient with primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Hematol Oncol Stem Cell Ther. 2021; 14: 71-75.