
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Juvenile antisynthetase syndrome successfully treated by rituximab
Forough Emami1; Reza Shiari1*; Mohammad Samami2; Sara Shiari3; Yalda Nilipour4; Khosro Rahmani1; Azadeh Zeinab Mirzaee3; Vadood Javadi Parvaneh1; Niloofar Shashaani1; Reza Sinaei5,6
1Shahid Beheshti University of Medical Sciences, Department of Pediatric Rheumatology, Tehran, Iran.
2Dental Science Research Center, Department of Oral and Maxillofacial Medicine, Guilan University of Medical Sciences, Rasht, Iran.
3Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Pediatric Respiratory Diseases Research Center (PRDRC), Masih Daneshvari Hospital, Tehran, Iran.
4Pediatric Pathology Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
5Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Pediatric Respiratory Diseases Research Center (PRDRC), Masih Daneshvari Hospital, Tehran, Iran.
6Department of Pediatrics, School of Medicine, Kerman University of Medical Sciences, Kerman, Iran.
*Corresponding Author : Reza Shiari
Professor in Pediatric Rheumatology, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Tel: +98-21-22227033;
Email: Shiareza@yahoo.com
Received : Oct 10, 2023
Accepted : Nov 09, 2023
Published : Nov 16, 2023
Archived : www.jcimcr.org
Copyright : © Shiari R (2023).
Abstract
Background: The Antisynthetase Syndrome (ASS) is a rare autoimmune disease considered as a subset of idiopathic inflammatory myopathies. This syndrome is associated with antisynthetase antibodies and clinical manifestations such as Raynaud’s phenomenon, mechanic’s hands and interstitial lung disease.
Case presentation: Here in we reported a 16 years old Afghani girl who was diagnosed as having ASS. Rare diseases such as ASS should be suspected in idiopathic inflammatory myopathy patients who are resistant to standard medical regimens.
Conclusion: In such cases, the use of Rituximab is strongly recommended. To the best of our knowledge is the first report of juvenile ASS in Middle East.
Keywords: Antisynthetase syndrome; Myopathy; Rituximab; Autoimmune disease.
Abbreviations: ASS: Antisynthetase Syndrome; IIM: Idiopathic Inflammatory Myopathies; ILD: Interstitial Lung Disease; AST; Aspartate Aminotransferase; ALT: Alanine Aminotransferase; CPK: Creatine Phosphokinase; LD: Lactate Dehydrogenize; ANA: Antinuclear Antibody; CRP: C-Reactive Protein; ESR: Erythrocyte Sedimentation Rate; EMG: Electromyography; NCV: Nerve Conduction Velocity; HRCT: High Resolution Computed Tomography; IVIG: Intravenous Immunoglobulin; JDM: Juvenile Dermatomyositis; PM: Polymyositis; DM: Dermatomyositis; MTX: Methotrexate.
Citation: Emami F, Shiari R, Samami M, Shiari S, Nilipour Y, et al. Juvenile antisynthetase syndrome successfully treated by rituximab. J Clin Images Med Case Rep. 2023; 4(11): 2691.
Introduction
Myopathies are classified into two main categories: inflammatory and non-inflammatory [1,2]. The Antisynthetase Syndrome (ASS) is a rare autoimmune connective tissue disease defines as a subset of Idiopathic Inflammatory Myopathies (IIM) associated with antisynthetase antibodies and various clinical manifestations such as Raynaud’s phenomenon, mechanic’s hands and Interstitial Lung Disease (ILD) [3,4]. Here in we reported a 16-year-old Afghan female with 3 months history of muscle weakness but no previous history of systemic disease who was finally diagnosed as antisynthetase syndrome. To the best of our knowledge this is the first report of juvenile ASS from Middle East.
Case report
A 16-year-old Afghani girl with no record of any systemic disease, presented with a 3 months history of muscle weakness especially in proximal upper and lower extremities, positive gower sign and fatigue. Furthermore, she had a 1 month history of Raynaud’s phenomenon, erythema of extensor surfaces of hands, bilateral hand and foot swelling, periorbital edema, skin photosensitivity and diffuses labial swelling. Dyspnea on exertion and dysphagia were occurred later. In more evaluation, nail fold Telangiectasias was detected.
Initially, baseline studies were done. laboratory studies revealed elevated aspartate Aminotransferase (AST) of 252 U/L, Alanine Aminotransferase (ALT) of 162 U/L, Creatine PhosphoKinase (CPK) of 5773 IU/L, Lactate Dehydrogenize (LDH) of 1772 U/L, negative ANA(1:40), C-Reactive Protein (CRP) of 2 mg/L, Erythrocyte Sedimentation Rate (ESR) of 68 mm/h. Electromyography (EMG) and Nerve Conduction Velocity (NCV) tests concluded that was consistent with an irritative generalized symmetric myopathic disorder with evidence of active denervation.
Imaging studies were accomplished including High Resolution Computed Tomography (HRCT) of chest, echocardiography and barium swallowing test. HRCT showed multifocal sub-pleural ground-glass lesions. The COVID-19 real time-PCR and antibodies were negative. Echocardiogram revealed mild pulmonary hypertension with borderline pulmonary arterial pressure.
A muscle biopsy and auto-antibodies measurements were performed. Although, treatment was started with single dose of Intravenous Immunoglobulin (IVIG) at 2 g/kg and followed by intravenous injection of methylprednisolone, one gram daily for 5 days based on initial diagnosis of Juvenile Dermatomyositis (JDM). However, her muscle weakness and exertional dyspnea did not improve significantly. The results of other auto-antibodies including: Anti JO-1, Anti M2, Anti dsDNA, Anti SS/A, Anti SS/B, Anti RNP, Anti Scl-70, Anti PM-100; all were negative. Muscle biopsy revealed some perifascicular necrotic fibers with some perimysial fragmentation but no inflammation, compatible with histological diagnosis of ASS (Figure 1).
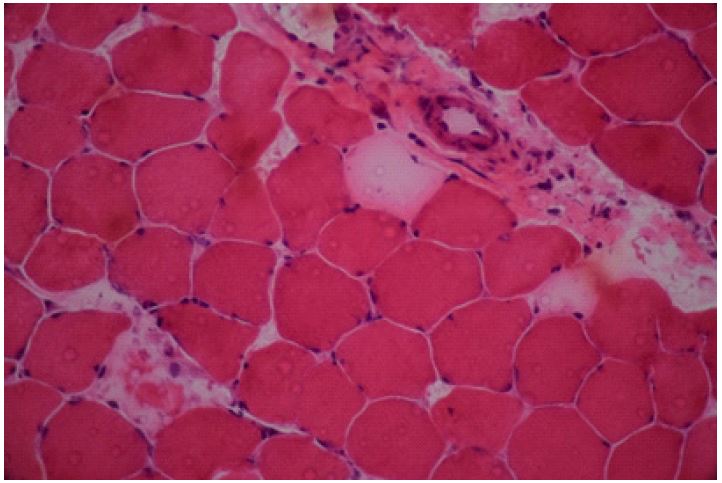
Perifascicular necrotic fibers with perimysial fragmentations, but no inflammation was detected (Haematoxylin and Eosin x400).
Following the ASS diagnosis, treatment with Rituximab at 375 mg/m2/week for 4 weeks was initiated. 2 months after starting the drug, the patient’s clinical signs and symptoms such as muscle weakness, exertional dyspnea, skin and nail manifestations, and dysphagi all relieved dramatically. Muscle enzymes and inflammatory markers were normalized. Amusingly, her ground-glass lesions of chest HRCT disappeared after 4 months. There were no signs or symptoms of the disease flare-up after one-year follow-up.
Discussion
ASS is a chronic connective tissue disease of unknown cause. Three sub-groups of IIMs include: 1) sporadic inclusion-body myositis; 2) polymyositis (PM); and 3) dermatomyositis (DM). ASS is a sub-class of PM/DM [5]. This chronic disease was first described by Marguerie et al. in 1990 [6]. The pathogenesis of this disease is explained by the presence of myositis-specific autoantibodies against aminoacyl tRNA synthetase, which may occur due to genetic predisposition, viral agents or medications. The most common autoantibody is anti-JO-1 antibody which is present in 80% of ASS patients [2,7]. However, in our patient, the results of myositis-specific autoantibodies (Anti JO-1, Anti M2) and myositis-associated autoantibodies (Anti SS/A, Anti SS/B, Anti RNP, Anti Scl-70, Anti PM-100) all were negative.
Considering the rarity and delay in diagnosing of ASS, there is little data about its incidence and prevalence in the literature. The annual incidence of IIM and ASS in the general population has been reported approximately 2 and 0.6 per 100,000 individuals, respectively [8,9]. The disease primarily affects adults aged 22-74 years with an average age of about 50 years [5,8,10,11]. It develops in children extremely rare, with annual incidence of 2.5-4 cases per million and a peak incidence of 5-10 years old [12]. The females are more affected than men (2-3 ratio), and there is a predominance of caucasians [5,8,10-12]. Our case is a 17-year-old girl who, to the best of our knowledge, is the first report of juvenile ASS from the Middle East.
Clinical manifestations of the disease can include fever, fatigue, ILD, inflammatory myopathy, non-erosive arthritis; arthralgia, skin manifestations such as mechanic’s hands, Raynaud’s phenomenon and oral manifestations such as dry mouth and ulcers [7,10].
Our patient had fatigue, muscle weakness, Raynaud’s phenomenon, hand erythema, bilateral swelling and arthralgia of the hands and feet, orofacial edema, photosensitivity, exertional dyspnea and dysphagia. Inflammatory myopathy was confirmed by performing EMG-NCV.
The patient also had borderline PAP and ILD, which was diagnostic on the chest HRCT as ground-glass opacities [10]. It is commonly seen in the adults and has been reported rarely in children [13]. ILD overall prevalence has been reported to be 60%-100% according to various studies, since it occurs in up to 30% of patients as an initial presentation [5,14,15]. It is considered the most important prognostic factor (predicting morbidity and mortality) [16,17] which manifests as exertional dyspnea and dry cough. ILD may cause pulmonary hypertension, however, pulmonary hypertension development without ILD has also been reported [5,14]. Anti-JO-1 antibodies have been reported as the most important predictor of ILD in patients with ASS and is detectable in 60-70% of ASS patients with ILD [5,18].
Because of low prevalence of ASS and lack of adequate randomized clinical trials, the treatment of these patients is challenging. However, treatment of this kind of IIM is typically based on the severity of ILD or myositis [14,19]. The most common recommended initial medication is systemic corticosteroids (prednisone or methylprednisolone). Depending on the severity of the disease, a steroid sparing agent such as azathioprine can be added which decreases the rates of relapses and improves survival [14,18]. Other immunomodulating agents such as IVIG, Methotrexate (MTX), cyclophosphamide and cyclosporine have also been mentioned as effective medications in various studies. IVIG is used to treat myositis in IIM, but is not effective in cases of ILD [15,20]. Recently, there has been a growing tendency to use of Rituximab in the treatment of ASS specifically for patients with refractory ILD and/or myositis [19]. According to studies, this drug can cause significant improvement in clinical symptoms and imaging, especially in patients with duration of less than one year [14,15,20]. In current reported case, the patient was initially treated with IVIG and intravenous methylprednisolone pulse which did not reviled symptoms. In the second line, the patient was treated with Rituximab and showed dramatic improvement in symptoms after 2 months. HRCT imaging manifestations of the ILD disappeared entirely within 4 months, during which no evidence of infection was found. Maintenance treatment was continued with oral prednisolone tapering and subcutaneous MTX. There is no evidence of muscle weakness, ILD or any relapsing symptoms during the 9 months follow-up.
Conclusion
Rare diseases such as antisynthetase syndrome must are suspected in idiopathic inflammatory myopathy patients who are resistant to standard treatment. In such cases, the use of Rituximab is strongly recommended.
References
- Manole E, Bastian AE, Butoianu N, Goebel HH. Myositis non-inflammatory mechanisms: An up-dated review. Journal of Immunoassay and Immunochemistry. 2017; 38: 115-26.
- Chatterjee S, Prayson R, Farver C. Antisynthetase syndrome: Not just an inflammatory myopathy. Cleveland Clinic Journal of Medicine. 2013; 80: 655-66.
- Gasparotto M, Gatto M, Saccon F, Ghirardello A, Iaccarino L, et al. Pulmonary involvement in antisynthetase syndrome. Current opinion in rheumatology. 2019; 31: 603-10.
- Ponce-Gallegos MA, Ramos-Martínez E, García-Carmona A, Mejía M, Nava-Quiroz KJ, et al. Genetic susceptibility to Antisynthetase Syndrome associated with single-nucleotide variants in the IL1B gene that lead variation in IL-1β serum levels. Frontiers in medicine. 2020; 7.
- Badshah A, Haider I, Pervez S, Humayun M. Antisynthetase syndrome presenting as interstitial lung disease: a case report. Journal of medical case reports. 2019; 13: 1-6.
- Marguerie C, Bunn C, Beynon H, Bernstein R, Hughes J, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. QJM: An International Journal of Medicine. 1990; 77: 1019-38.
- Gormley M, Scully C. Antisynthetase syndrome: Two cases presenting orofacial manifestations. British Journal of Oral and Maxillofacial Surgery. 2014; 52: 285-7.
- Marin FL, Sampaio HP. Antisynthetase syndrome and autoantibodies: A literature review and report of 4 cases. The American journal of case reports. 2019; 20: 1094.
- Jensen ML, Løkke A, Hilberg O, Hyldgaard C, Tran D, et al. Clinical characteristics and outcome in patients with antisynthetase syndrome associated interstitial lung disease: A retrospective cohort study. European clinical respiratory journal. 2019; 6: 1583516.
- Baccaro A, Pinto GB, Carboni R, Shinjo S. The clinical manifestations at the onset of antisynthetase syndrome: a chameleon with multiple faces. Reumatismo. 2020; 72: 86-92.
- Boleto G, Perotin JM, Eschard JP, Salmon JH. Squamous cell carcinoma of the lung associated with anti-Jo1 antisynthetase syndrome: A case report and review of the literature. Rheumatology international. 2017; 37: 1203-6.
- Asi K, Gourishankar A, Kamdar A. Coronary artery dilation associated with anti-synthetase syndrome in an adolescent. Pediatric Rheumatology. 2019; 17: 1-3.
- Hayes D, Baker PB, Mansour HM, Peeples ME, Nicol KK. Interstitial lung disease in a child with antisynthetase syndrome. Lung. 2013; 191: 441-3.
- Alfraji N, Mazahir U, Chaudhri M, Miskoff J. Anti-synthetase syndrome: A rare and challenging diagnosis for bilateral ground-glass opacities-a case report with literature review. BMC pulmonary medicine. 2021; 21: 1-6.
- Marco JL, Collins BF. Clinical manifestations and treatment of antisynthetase syndrome. Best Practice & Research Clinical Rheumatology. 2020; 34: 101503.
- García-Fernández A, Quezada-Loaiza CA, de la Puente-Bujidos C. Antisynthetase syndrome and pulmonary hypertension: Report of two cases and review of the literature. Modern Rheumatology Case Reports. 2021; 5: 152-5.
- Cancel M, Song M. Acute hypoxic respiratory failure secondary to antisynthetase syndrome: A case report and review of literature. Respiratory medicine case reports. 2019; 26: 288-91.
- Huang K, Aggarwal R. Antisynthetase syndrome: A distinct disease spectrum. Journal of Scleroderma and Related Disorders. 2020; 5: 178-91.
- Cherin P, de Jaeger C, Crave JC, Delain JC, Tadmouri A, et al. Subcutaneous immunoglobulins for the treatment of a patient with antisynthetase syndrome and secondary chronic immunodeficiency after anti-CD20 treatment: a case report. Journal of medical case reports. 2017; 11: 1-6.
- Zanframundo G, Marasco E, La Carrubba C, De Stefano L, Volpiano L, et al. Update on Treatment of Antisynthetase Syndrome: A Brief Review. Current Treatment Options in Rheumatology. 2020; 6: 18-28.