
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 4
Digital gangrene: A rare harbinger of Waldenström’s macroglobulinemia
*Corresponding Author : Faheema Hasan
Assistant Professor, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India.
Email: 1faheemahasan@gmail.com
Received : Oct 27, 2023
Accepted : Nov 22, 2023
Published : Nov 29, 2023
Archived : www.jcimcr.org
Copyright : © Hasan F (2023).
Abstract
Waldenström’s Macroglobulinemia (WM) is a rare lymphoma characterized by monoclonal Immunoglobulin M (IgM) production and can remain asymptomatic for extended periods. Cryoglobulinemia is a known manifestation of WM, but cryoglobulinemic vasculitis causing digital gangrene as the initial presentation is exceedingly rare. We describe the case of a 40-year-old female who presented with cryoglobulinemic vasculitis-induced dry gangrene of her toes and fingers ten months before the diagnosis of WM. Digital gangrene in the absence of other clinical features pertaining to WM is unusual and should prompt consideration of underlying lymphoproliferative disorders. This case emphasizes the importance of considering underlying B-cell malignancies in the differential diagnosis of digital gangrene and the possibility that cutaneous manifestations may precede other disease manifestations by months or years.
Keywords: Monoclonal immunoglobulins; Blood hyperviscosity; Blood hyperviscosity; Plasma cell dyscrasias; Coldagglutinin; Symmetrical peripheral gangrene (spg); Waldenstrom macroglobulinemia.
Citation: Hasan F. Digital gangrene: A rare harbinger of Waldenström’s macroglobulinemia. J Clin Images Med Case Rep. 2023; 4(11): 2713.
Introduction
Waldenstrom’s Macroglobulinemia (WM) is a rare and indolent lymphoma characterised by infiltration of Bone Marrow (BM) by lymphoplasmacytic cells and production of monoclonal Immunoglobulin M (IgM). The median age of presentation is 63 years and is more common in males. It can remain asymptomatic for years as IgM MGUS (monoclonal gammopathy of undetermined significance) or as smoldering macroglobulinemia [1]. When symptomatic, the manifestations are related to the IgM monoclonal protein and/or progressive BM and nodal infiltration with lymphoma cells [2]. Cryoglobulinemia is an uncommon manifestation of Waldenstrom’s Macroglobulinemia and can be seen in up to 1% of patients at presentation [1-3]. However, cryoglobulinemic vasculitis causing digital gangrene and heralding the diagnosis of WM by several months has not been reported before. We present a case of a 40-year-old female who presented with cryoglobulinemic vasculitis induced dry gangrene of toes and fingers ten months before the diagnosis of WM.
Case presentation
A 40-year-old female presented with blackish discoloration of distal toes in both feet, multiple blackish discoloration of fingers of both hands along with pain and puffiness in fingers for five days. There was no history of fever, chest pain, visual blurring, headache, breathlessness, hematuria/oliguria, photosensitivity, arthralgia, Raynaud’s symptoms or sicca symptoms. Examination revealed dry gangrene of bilateral toes and black patches in fingers, however all peripheral pulses were palpable and there was no lymphadenopathy or organomegaly (Figure 1).
Baseline investigations revealed Hb: 10.2 gm%, Total Leucocyte count of 8,800/cu mm with 90% polymorphs and 10% lymphocytes and a platelet count of 1.37 Lacs/cu mm. The Erythrocyte Sedimentation Rate (ESR) was 70 mm/hr. Liver function, renal function and coagulation assays were within normal limits. Ultra sound doppler revealed thrombosis of bilateral anterior tibial veins, but no arterial thrombosis. Anti Nuclear Antibody/ Anti Neutrophil Cytoplasmic Antibodies/Beta 2 GP1 Antibodies /Lupus Anticoagulant/Cryoglobulins were negative. Ultrasound of the whole abdomen was performed and was normal. Patient was started on aspirin and heparin and switched to warfarin with bridging. APLA screening was repeated in six weeks and still was negative. Gangrenous toes spontaneously shed off with time and puffiness and pain of the fingers also resolved. Anticoagulation was advised to be stopped after six months as no cause was found.
She was on regular follow up in the immunology OPD. However, ten months after the initial episode, she developed global headache followed by loss of consciousness after a fall. She was evaluated at a local hospital where she was diagnosed to be having acute Sub Dural Hematoma and was treated conservatively with anti-oedema measures after which she regained consciousness. She was then transferred to our centre. She also complained of early satiety, abdominal fullness, malaise and easy fatiguability for 3 months. On examination, she was conscious, oriented, hemodynamically stable. She had pallor, no palpable lymphadenopathy but massive Splenomegaly extending till the iliac fossa and hepatomegaly measuring 4 cms below the costal margin. This time the hemogram revealed Hb: 6.6 gm/dl, Total Leucocyte Count of 4400/cu mm with 53% polymorphs and 47% lymphocytes and a platelet count of 0.77 Lacs/cmm. The ESR was 122 mm/hr. Liver function , renal function and coagulation assays were within normal limits. Bone marrow aspiration and biopsy was planned in view of splenomegaly. Marrow examination revealed increased lymphocytes (25-30%) and increased plasmacytoid cells (8-10%) on aspiration and the biopsy showed clusters of atypical lymphoid cells (Figure 2). Flowcytometry showed that these cells are CLPD CD5- /CD10-, CD200+, FMC7 dim positive consistent with a diagnosis of Waldenstrom’s Macroglobulinemia (Figure 3). Serum Protein Electrophoresis revealed an M band of 2.69 g/dl with immunofixation typing it to be IgM kappa monoclonal protein. Serum Free kappa light chain was 107 mg/l, Free lambda light chain was 15.8 mg/L with a kappa:lambda ratio of 6.7. IgM level was 6540 g/dl. Cold agglutinin assay was negative. Cryoglobulin was positive with a plasma viscosity of 2.8 cP. Autoantibodies, complement, viral markers were all negative.
Based on the above investigations, patient was diagnosed as a case of Waldenstrom’s Macroglobulinemia (WM) with type I cryoglobulinemia. She was planned for BDR regimen (Bortezomib, Dexamethasone, Rituximab) therapy and Rituximab was to be added later after IgM < 4 g/dl. However after 4 weeks of Bortezomib and Dexamethasone and on OPD follow up, she presented with altered sensorium for 2 days. On evaluation, her IgM levels had increased to 9.2 g and plasma viscosity was 6.5 cP. She was started on plasmapheresis and received 6 cycles of the same and became symptomatically better, IgM levels dropped to 3.6 G/dl and was started on Rituximab, Cyclophosphamide and Dexamethasone (RCD) therapy. Patient has received 8 cycles of RCD, and was then taken on Bortezomib maintenance. She is currently asymptomatic and is in remission with no detectable M protein for the past 2 years.

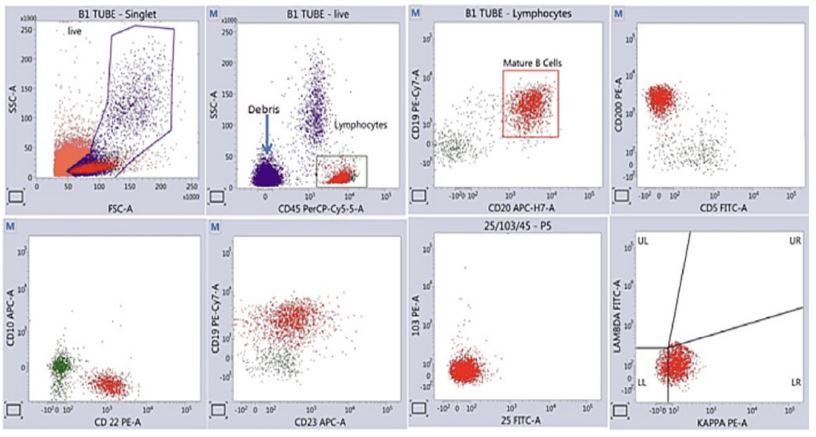
Discussion
Waldenström Macroglobulinemia (WM) is an uncommon Non-Hodgkin Lymphoma accounting for 1%-2% of hematological malignancies. It is characterized by the accumulation of lymphoplasmacytic cells that produce monoclonal immunoglobulin M [4]. The clinical features of WM are due to tumour burden or quantity, physicochemical or immunological properties of the monoclonal IgM. Around 19% to 28% of patients have asymptomatic disease and can remain asymptomatic for several years; median time to symptom development may exceed 5 years.
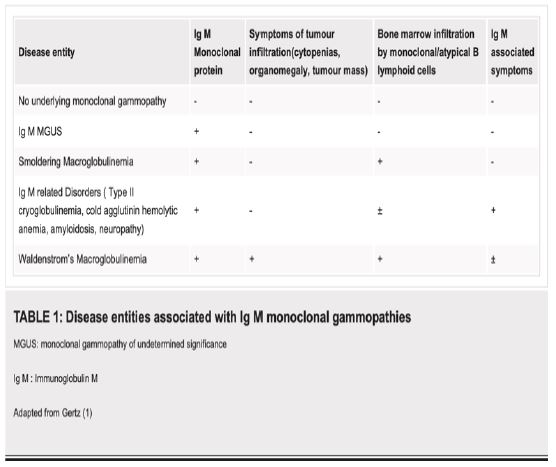
Presence of a IgM monoclonal protein without any symptoms of tumour mass or infiltration, IgM mediated symptoms or bone marrow involvement by >10% monoclonal B cells is called IgM MGUS (monoclonal gammopathy of undetermined significance), while in the presence of marrow infiltration and absence of symptoms, the term smoldering macroglobulinemia is used (Table 1) [1].
Majority of the patients with WM present with disease restricted to their bone marrow leading to cytopenias; especially anemia due to replacement of hematopoietic elements by tumour cells. While 5.5% of patients with symptomatic WM can have cryoglobulins, only 1.3% of patients with WM have cryoglobulins at initial presentation according to the Greek Myeloma data base. In our case, the patient presented 11 months preceding the diagnosis of WM with features of cryoglobulinemia. Cryoglobulins are immunoglobulins that precipitate below 37°C and redissolve on warming in vitro [5]. It was first identified by Wintrobe and Buell in 1933 in a 56 year old woman patient. Raynaud’s phenomenon and myeloma and was later termed cryoglobulins in 1947. Three classes of cryoglobulins are recognised. Type I cryoglobulins are monoclonal IgM, IgG or IgA and is seen in association with haematological malignancies like including WM, Multiple Myeloma (MM), Chronic Lymphocytic Leukemia (CLL) in 60% of the cases and the Monoclonal gammopathy of undetermined significance (MGUS) in 40%. Type II cryoglobulins are usually Monoclonal IgM, mixed with other polyclonal immunoglobulins are commonly associated with underlying viral infections, usually hepatitis C infection in up to 90% [5].
Type III cryoglobulins are mixed immunoglobulins composed of polyclonal IgM and IgG and is seen mostly with connective tissue disease and has polyclonal immunoglobulin precipitates. Type 1 cryoglobulins usually present with skin manifestations in up to 86% of patients. These include livedo reticularis, purpura, Raynaud’s phenomenon, acrocyanosis, skin necrosis, ulcers and, rarely, digital gangrene and are often confined to acral sites and are triggered by cold exposure. However, clinical manifestations are present during the time of diagnosis and are often accompanied by other features of the underlying haematological malignancy [2].
Our patient presented with digital gangrene with no other identifiable cause despite extensive investigations. Hemogram only showed mild anemia with HB of 10 gm%, autoantibody screening including for rheumatoid factor was negative, hepatitis B and C serology were negative and abdominal ultrasonogram was normal. At this time point cryoglobulins were also negative and serum protein electrophoresis was not done. At that time point, she could have harboured this IgM related disorder and the routine tests for cryoglobulinemia which has a lot of confounding factors due to loss of cryoprecipitate during storage and transport might have caused the diagnosis to be missed.
In the second visit, eleven months later, her anemia had worsened, possibly related to the underlying monoclonal gammopathy with increasing clone size evident by relative lymphocytosis and evidence of tumour infiltration in the form of massive splenomegaly which prompted the work up for an underlying lymphoproliferative disorder. Bone marrow examination, immunophenotyping and presence of IgM monoclonal band clenched the diagnosis of WM. Unfortunately, our centre was not equipped to do MYD 88 assay at that point of time .Our case is noteworthy because the type I cryoglobulinemic vasculitis was fore runner of WM by 11 months. There are only mere anecdotal reports of digital gangrene as the presenting feature of WM and MM, but on an extensive literature search, to the best of our knowledge, we could not find any case reports of type I cryoglobulinemic vasculitis heralding the diagnosis of WM by months. She responded very well to RCD chemotherapy followed by bortezomib maintenance for 2 years and is now on OPD follow up with no other issues. 30-70% of patients have reported a transient increase in IgM levels (flare) after rituximab therapy. Given the rise in IgM, plasmapheresis should be considered prior to rituximab if serum viscosity is greater than 3.5 cp or the IgM level greater than 5 g/dL. The use of combination chemotherapy regimens with rituximab may lessen the flare phenomenon [6].
Conclusion
This case is very unique because digital gangrene and skin infarcts were the sole presenting manifestation of WM and preceded the onset of the other classical clinical features of WM by almost a year. It highlights that while a number of common causes like autoimmunity, infections should be worked up in a case of digital gangrene, the possibility of an underlying B cell malignancy should always be considered a differential diagnosis and a simple work up like serum protein electrophoresis could identify a possible underlying monoclonal gammopathy.
References
- Morie A. Gertz. Waldenström macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. Annual Clinical Updates In Hematological Malignancies. 2021; 96: 258-269.
- Stone MJ, Pascual V. Pathophysiology of Waldenström’s macroglobulinemia. Haematologica. 2010; 3: 359-64.
- Ramos-Casals M, Stone JH, Cid MC, Bosch X. The cryoglobulinaemias. Lancet. 2012; 28: 348-60.
- Treon SP, Dimopoulos M, Kyle RA. Defining Waldenstrom’s macroglobulinemia. Semin Oncol. 2003; 30: 107-9.
- Morie A. Gertz; Cold Hemolytic Syndrome. Hematology Am Soc Hematol Educ Program. 2006; 1: 19-23.
- Stone MJ. Waldenström’s macroglobulinemia: Hyperviscosity syndrome and cryoglobulinemia. Clin Lymphoma Myeloma. 2009; 1: 97-9.