
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Decisive role of mRNA sequencing to solve a suspicion of Fabry-disease in a patient with hypertrophic cardiomyopathy and normal GLA DNA sequencing
Albain Chansavang1*; Flavie Ader1,2,3*; Foudil Lamari4; Caroline Rooryck5; Françoise Pousset6; Julie Proukhnitzky2,6; Céline Bordet6; Jean-Pierre Rabes7; Philippe Charron2,6#; Pascale Richard1,2#
1APHP, Pitié Salpêtrière- Sorbonne University, DMU BioGem, UF Cardiogenetics and Molecular and Cellular Myogenetics, Department of Metabolic Biochemistry, F-75013 Paris, France.
2Sorbonne University, INSERM UMRS 1166 and ICAN Institute, F-75013, Paris, France.
3UFR Pharmacy University of Paris Cité, Department 3, F-75006 Paris, France.Complex
4APHP, Pitié Salpêtrière- Sorbonne University, DMU BioGem, UF Neurometabolism, Department of Metabolic Biochemistry, Paris, F-75013 France.
5Bordeaux University Hospital, Medical Genetics Department, F-33000 Bordeaux, France.
6APHP, Reference Center for Hereditary or Rare Heart Diseases, Department of Genetics and Department of Cardiology, Pitié Salpêtrière Hospital, Paris, F-75013 France.
7APHP, Biochemistry and Genetics Laboratory, Ambroise Paré Hospital, Boulogne, France.
#Equal Last Authors.
*Corresponding Author : Flavie Ader
Cardiogenetics and Myogenetics Functional Unit, Genetics Center, Pharmacy Building, Pitié Salpêtrière University Hospitals - Charles Foix 47/83 boulevard of The Hospital 75013, Paris.
Tel +33-1-42-17-7656 & +33-1-42 17-76 18;
Email: flavie.ader@aphp.fr
Received : Dec 19, 2023
Accepted : Jan 09, 2024
Published : Jan 16, 2024
Archived : www.jcimcr.org
Copyright : © Ader F (2024).
Abstract
We report the case of a 44-year-old-male presenting with hypertrophic cardiomyopathy and a biochemical analysis suggestive of a Fabry-disease but with a negative coding-regions sequencing of GLA gene (Next Generation Sequencing (NGS), Sanger sequencing and GLA large rearrangement assay). GLA mRNA sequencing was subsequently performed and revealed an abnormal transcript with an intronic sequence insertion due to the deep-intronic hemizygous c.640-801G>A substitution. This deep-intronic region was not sequenced with the next generation custom panel used based on coding regions analysis. In the presence of a clinical suspicion of Fabry-disease and negative results of gene coding regions sequencing, transcript sequencing appears as an appropriate strategy that should be consider solving the diagnostic issue. Following this diagnosis, enzyme replacement therapy has been proposed to the patient, as well as targeted genetic analysis in the family in order to prevent disease progression in relatives.
This case illustrates a particular molecular pitfall regarding the diagnosis of some rare inherited diseases and limitations of NGS/Sanger strategies. Further sequencing mRNA of genes of interest could be a strategy in case of no diagnosis obtained through conventional NGS panel in cardiomyopathies.
Keywords: Fabry disease; RNA sequencing; Cardiomyopathy; lysoGB3.
Citation: Chansavang A, Ader F, Lamari F, Rooryck C, Pousset F, et al. Decisive role of mRNA sequencing to solve a suspicion of Fabry-disease in a patient with hypertrophic cardiomyopathy and normal GLA DNA sequencing. J Clin Images Med Case Rep. 2024; 5(1): 2798.
Introduction
Fabry disease (OMIM 301500) is an X-linked disorder caused by pathogenic variants in GLA gene (NM_000169.3; GenBank accession number NG_007119) leading to a reduction or absence of alpha-galactosidase activity and accumulation of upstream metabolites such as lyso-globotriaosylceramide (lyso-Gb3). In male patients, Fabry disease is divided into “classic” Fabry disease with absent alpha-galactosidase activity, a childhood or adolescent onset and a multiorgan failure at delayed onset, and the “delayed/incomplete” Fabry disease form, characterized by late-onset clinical manifestations during the fourth decade of life including hypertrophic cardiomyopathy and sometimes associated with a decreased glomerular filtration rate, due to residual alpha-galactosidase activity [1].
Confirmatory diagnosis of Fabry disease is usually made by a combination of alpha-galactosidase activity and GLA gene disease-causing variant identification, the latter is especially useful for subsequent genetic counseling/testing in relatives and also for the therapeutic discussion since some specific treatments are possible only for particular GLA variants [1].
Case presentation
Here, we report the case of a 44-year-old male patient from Cambodia presenting hypertrophic cardiomyopathy (HCM) (Interventricular septum thickness: 30 mm) and slightly reduced glomerular filtration rate (50 mL/min/1.73 m2 CKD-EPI formula, and moderate proteinuria 0,11 g/24 hours). No cause of chronic kidney disease at this point has been identified, except history of blood hypertension but well controlled by therapy for many years. No stroke or neuropathic pain were reported. Echocardiography (TTE) revealed Left Ventricular (LV) hypertrophy with maximal wall thickness 30 mm, LV Ejection Fraction (EF) 50% and no outflow tract gradient. The cardiac magnetic resonance imaging (MRI) revealed important late enhancement (diffuse multifocal enhancement >15% LV mass) but without remarkable regional localization and normal T1 mapping relaxation time (1016 ms); LV ED Volume was 105 ml/m2, LV EF 60%, LV mass 143 g/m2. EKG exhibited a sinus rhythm, PR interval was 190 ms and complete LBBB (QRS duration 140 ms). On holter ECG, non sustained VT as well as few supraventricular bursts were recorded. Cardiac biomarkers were slightly increased at diagnosis (highly sensitive troponine=60 ng/l, normal range < 14 ng/l; and NTproBNP 462 pg/mL, normal range < 400 pg/mL).
However, leucocyte alpha-galactosidase activity was reduced to 13 nmol/h/mg (normal range 20-80 nmol/h/mg) and lyso-Gb3 was accumulated in plasma (10.1 nmol/L, normal range < 0.8 nmol/L) confirmed on several dosages. The patient has signed an informed written consent for genetic analysis. Genetic testing by Sanger sequencing for most frequent hypertrophic cardiomyopathy sarcomeric genes (MYH7, MYBPC3, TNNT2, TNNI3), TTR and GLA failed to detect any pathogenic variant. GLA intragenic deletions or duplications were ruled out using by oligonucleotides 60K Custom arrays (Agilent Technologies, one probe every 100 pb), and high throughput sequencing of a panel of 71 genes implicated in cardiomyopathy (including GLA) did not detect any pathogenic variant in coding and flanking intronic sequences. After the first negative molecular investigations, further clinical examination did not reveal angiokeratoma but revealed cornea verticillata. In front of the evocative diagnosis of Fabry disease, analysis of GLA mRNA was performed from the patient lymphoblastic cells after reverse transcription and cDNA sequencing. This analysis revealed an abnormal GLA transcript containing a 57 bp pseudo-exon inclusion. Genomic DNA sequencing of the corresponding intronic region identified the presence of a hemizygous GLA substitution variant NM_000169.3:c.640-801G>A (also named IVS4+919G>A). This variant disrupts a silencer sequence, removing the splicing inhibition resulting in the pseudo-exon inclusion leading to a non-functional protein [2]. Following this diagnosis, enzyme replacement therapy has been proposed to the patient (1.0 mg /kg of Agalsidase alpha). Additionally, targeted genetic analysis have been performed in the family in order to prevent adapt cardiological follow up in the carriers (hemizygous males as well as heterozygous females).
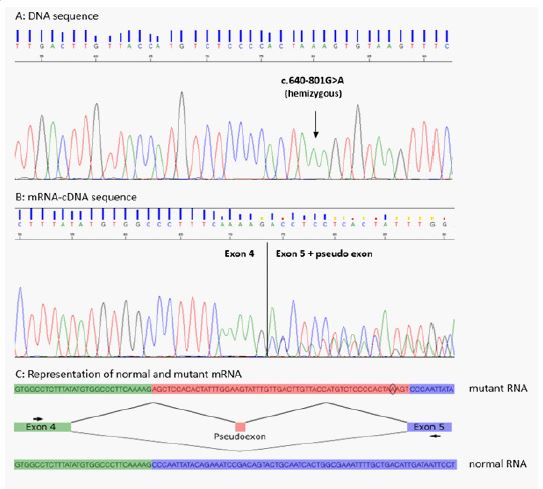
(A) Sanger sequencing in the proband of the GLA intron 4 region. The GLA genomic variant located position -801 from the exon 5 junction is indicated by an arrow. (B) Electrophoregram of mRNA sequencing of GLA region (exon 4 and exon 5) including the pseudo exon. The insertion of the pseudo exon is visualized by the superposed sequences. (C) Schematic representation of the mechanism leading to the insertion of the pseudo exon: Green rectangle represents exon 4, purple rectangle represents exon 5 and red rectangle represents the intronic regions corresponding to the pseudo-exon.
Discussion and conclusion
We report the case of male patient presenting a late-onset Fabry disease with a moderately reduced alpha-galactosidase activity and increased lyso-Gb3 level. Both Sanger and high throughput coding DNA sequencing failed to identify the pathogenic variant but GLA transcript analysis revealed the presence of a deep intronic pathogenic variant (NM_000169.3:c.640-801G>A) disrupting normal splicing of GLA gene.
This pathogenic variant was previously described in Japanese late-onset Fabry disease male patients presenting 8-15% of residual enzyme activity [2-4] and functional experiments confirmed consequences at protein level [2]. Newborn screening for late-onset Fabry disease variants found that this variant was absent of Korean newborns [5] but highly prevalent in male Taiwanese newborns (1/1460 – 1/875) with < 30% of residual enzyme activity [6,7]. The geographic origin of the patient is consistent with the previous cases described. The phenotype associated with this variant in adult male is described to vary from no left ventricular hypertrophy to severe concentric hypertrophy [8], and the expression of the disease didn’t seem to be linked to additional pathogenic/likely pathogenic variant [9]. In our patient, the sequencing of coding region of 71 cardiomyopathies genes did not reveal the presence of additional pathogenic/likely pathogenic variant. Familial segregation has also been performed within the family (3 women and 1 man of 50 years old, all asymptomatic with normal cardiological examination EKG and TTE) which is consistent with this phenotypic heterogeneity.
This report first emphasizes the importance of alpha-galactosidase and lyso-Gb3 dosages in front of isolated HCM to properly rule out or suspect Fabry disease in male patients. Second it shows that DNA coding regions sequencing may fail identifying the underlying pathogenic variant especially in case of deep intronic variants. Therefore, a moderately lowered residual enzyme activity observed in a male patient and a negative DNA coding sequencing should not rule out Fabry diagnosis in patients with a clinical/biochemical suspicion of Fabry disease confirming the importance of an accurate clinical diagnosis. Third, this report shows the crucial role of GLA transcript (mRNA) analysis in front of a suggestive diagnosis of Fabry disease for subsequent genetic counseling/testing in relatives and for the putative therapeutic discussion. Thus, this region should be considered in the NGS panel and the Sanger sequencing on DNA in order to improve efficiency of molecular diagnosis of Fabry cases. In conclusion, the medical community should be aware of such particular molecular pitfalls regarding the diagnosis of rare inherited diseases, and the interest of direct transcript analysis.
Conflict of interest: None declared.
Funding: This work was supported by Aviesan-ITMO Genetique-Genomique-Bioinformatique [ResDiCard: Resolving diagnostic deadlock in cardiomyopathies] and Federation Francaise de Cardiologie (P. Richard Grant).
References
- Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018; 123: 416-27.
- Palhais B, Dembic M, Sabaratnam R, Nielsen KS, Doktor TK, et al. The prevalent deep intronic c. 639+919 G>A GLA mutation causes pseudoexon activation and Fabry disease by abolishing the binding of hnRNPA1 and hnRNP A2/B1 to a splicing silencer. Mol Genet Metab. 2016; 119: 258-69.
- Ishii S, Nakao S, Minamikawa-Tachino R, Desnick RJ, Fan J-Q. Alternative splicing in the alpha-galactosidase a gene: Increased exon inclusion results in the Fabry cardiac phenotype. Am J Hum Genet. 2002; 70: 994-1002.
- Kobayashi M, Ohashi T, Kaneshiro E, Higuchi T, Ida H. Mutation spectrum of α-Galactosidase gene in Japanese patients with Fabry disease. J Hum Genet. 2019; 64: 695-9.
- Choi JH, Lee BH, Heo SH, Kim GH, Kim YM, et al. Clinical characteristics and mutation spectrum of GLA in Korean patients with Fabry disease by a nationwide survey: Underdiagnosis of late-onset phenotype. Medicine (Baltimore). 2017; 96: 7387.
- Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 2009; 30: 1397-405.
- Chien YH, Lee NC, Chiang SC, Desnick RJ, Hwu WL. Fabry disease: Incidence of the common later-onset α-galactosidase A IVS4+919G→A mutation in Taiwanese newborns--superiority of DNA-based to enzyme-based newborn screening for common mutations. Mol Med Camb Mass. 2012; 18: 780-4.
- Hsu MJ, Chang FP, Lu YH, Hung SC, Wang YC,et al. Identification of lysosomal and extralysosomal globotriaosylceramide (Gb3) accumulations before the occurrence of typical pathological changes in the endomyocardial biopsies of Fabry disease patients. Genet Med. 2019; 21: 224-232.
- Juang, JMJ, Shun CT, Chen YS, Hwu WL, Lee NC, et al. Fabry disease cardiac variant IVS4+919 G>A is associated with multiple cardiac gene variants in patients with severe cardiomyopathy and fatal arrhythmia. Genet Med. 2019; 21: 1890-1891.