
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
PRES syndrome in an adult patient with sickle cell disease and hyperhemolysis syndrome and review of the literature
Iliana Mani1; Aikaterini Xydaki2; Efrosyni Nomikou3; Olga Smyrniou3; Ioanna Segkou1; Alexandra Alexopoulou1*; Sophia Delicou2
12nd Department of Internal Medicine and Research Laboratory, Medical School, National & Kapodistrian University of Athens, Hippokration General Hospital, Greece.
2Thalassemia and Sickle Cell Unit, Expertise Center of Hemoglobinopathies and Their Complications, Hippokratio General Hospital of Athens, Athens, Greece.
*Corresponding Author : Alexandra Alexopoulou
2nd Department of Internal Medicine and Research Laboratory, Medical School, National & Kapodistrian University of Athens, Hippokration General Hospital, Vas. Sofias 114 Athens 11527, Greece.
Tel +30 213 2088129, Fax +30 210 7706871;
Email: alexopou@ath.forthnet.gr
ΟRCID ID: 0000-0003-1991-7436
Received : Dec 21, 2023
Accepted : Jan 09, 2024
Published : Jan 16, 2024
Archived : www.jcimcr.org
Copyright : © Alexopoulou A (2024).
Abstract
Sickle cell disease refers to a group of inherited disorders resulting from mutations in the gene encoding the haemoglobin subunit β. Main pathophysiological determinants of the disease’s course are vaso-occlusion, systemic inflammatory reaction and endothelial dysfunction. Phenotype is highly diverse, including acute and chronic complications of various organ systems, leading to significant morbidity and mortality. The natural course is further impacted by adverse reactions of therapeutic interventions, with transfusion reactions been among the most challenging to handle.
Here, we describe a patient with sickle cell disease with posterior reversible encephalopathy syndrome (PRES) as an unusual complication of hyperhemolysis syndrome, highlighting difficulties in diagnosis and management. We also performed a comprehensive literature review regarding this rare neurological complication in adult sickle cell disease.
Keywords: Sickle cell disease; Posterior reversible encephalopathy syndrome; Transfusion reaction; Alloantibodies; Hyperhemolysis.
Abbreviations: CT: Computed tomography; DHTRs: Delayed hemolytic transfusion reactions; PRES: Posterior reversible encephalopathy syndrome; RBCs: Red blood cells; SCD: Sickle cell disease.
Citation: Mani I, Xydaki A, Nomikou E, Smyrniou O, Alexopoulou A, et al. PRES syndrome in an adult patient with sickle cell disease and hyperhemolysis syndrome and review of the literature. J Clin Images Med Case Rep. 2024; 5(1): 2799.
Introduction
Sickle cell disease (SCD) is an inherited autosomal recessive blood disorder that affects the structure of hemoglobin and the shape of red blood cells (RBCs). A single mutation in the β-globin gene leads to the production of abnormal hemoglobin S, which polymerizes under low oxygen conditions, distorting RBCs into a sickle shape. These abnormal and rigid RBCs can obstruct blood vessels, impeding blood flow and causing tissue ischemia, inflammation, and multi-organ damage [1].
Common complications of SCD include painful vaso-occlusive crises, stroke, acute chest syndrome, splenic sequestration, chronic kidney disease, pulmonary hypertension, and early mortality [1]. Due to these severe complications, patients with SCD often require RBC transfusions to improve oxygen delivery and suppress endogenous sickle hemoglobin production. However, transfusions substantially increase the risk of complications like delayed hemolytic transfusion reactions (DHTRs) and alloimmunization in SCD patients compared to the general population [2].
Table 1: Main characteristic of cases of PRESS syndrome in adults with sickle cell disease.
| Author / year | Age, sex | Genotype | Presentation | Precipitatingfactor | Treatment | Outcome | Ref |
|---|---|---|---|---|---|---|---|
| Vargas A et al, 2019 | 25, F | NA | Seizures | Hypertension | NA | Resolution | 12 |
| Vargas A et al, 2019 | 34, F | NA | Seizures, visual changes | Hypertension, tacrolimus | NA | Resolution | 12 |
| Vargas A et al, 2019 | 30, M | NA | Seizures | Vaso-occlusive crisis | NA | Resolution | 12 |
| Vargas A et al, 2019 | 32, F | NA | Seizures | Hypertension | NA | Resolution | 13 |
| Raj S et al, 2013 | 19, F | SS | Headache, visual changes, seizures | Hypertransfusion | Phlebotomy | Resolution | 13 |
| Parameswaran BK et al, 2007 | 20, M | NA | Headache, visual changes confusion | Hypertension | Antihypertensivemedications | Resolution | 14 |
| Aiyer R et al, 2016 | 29, M | NA | Hemiplegia, lethargy, dysarthria, aphasia,facial droop | Acute chest syndrome, hypertension | Levetiracetam,phenytoin | Resolution | 15 |
| Geevasinga N, 2016 | 30, F | Sβ | Aphasia, pyramidal manifestations | Infection, ischemic stroke | Aspirin,antibiotics | Sepsis, death | 16 |
| Landais A et al, 2015 | 57, F | NA | Confusion, behavioral changes, right motordeficit, dysarthria | Vaso-occlusive crisis | NA | Resolution | 17 |
| Nair A et al, 2011 | 27, F | NA | Headache, seizures |
Acute pain crisis | Exchangetransfusion | Resolution | 18 |
| Sweany JM et al, 2007 | 56, F | NA | Seizures | Infection, hypertension, hemolysis | NA | Resolution | 19 |
| Zuccoli G et al, 2018 | 18, F | SS | Headache, visual changes, leg left weakness |
Subarachnoid hemorrhage, reversiblecerebral, vasoconstriction syndrome, hypertension | Exchange transfusion | 20 | |
| Present case | 40, F | Sβ | Coma | Hyperhaemolysis syndrome | Methylprednisolone,Gamma-globulin | Resolution |
This case report discusses two under-recognized transfusion risks that require heightened clinical vigilance in patients with SCD: 1) DHTRs, especially when accompanied by hyperhemolysis, and 2) posterior reversible encephalopathy syndrome (PRES). Prompt recognition and appropriate management of these complications are essential to reduce transfusion-related morbidity and mortality in SCD. We also conducted a review of the existing literature on the characteristics and outcomes of PRES syndrome in adult patients with sickle cell disease (Table 1).
Clinical findings
A 40-year-old woman diagnosed with Hb Sβ+-thalassemia, without proper medical monitoring, presented to the emergency department with a temperature of 39°C and backache. These symptoms had started 24 hours prior to admission. The patient had two hospital admissions within the last month for severe vaso-occlusive pain episodes, where she received IV antibiotics and three red blood cell simple transfusions. The most recent transfusion, conducted 10 days ago, increased her hemoglobin to 11.6 g/dl. At admission, the physical examination revealed nothing but fever and painless splenomegaly. Laboratory investigations showed anemia with hemoglobin levels at 7 g/dl, mean corpuscular volume (MCV) of 71.2 fl, mean corpuscular hemoglobin (MCH) of 24.2 pg, lactate dehydrogenase (LDH) of 1270 U/L (normal 125-220 U/L), total bilirubin of 6.98 mg/dl with direct bilirubin of 2.6 mg/dl, AST of 259 U/L (normal 5-34 U/L), ALT 225 U/L (normal 0-55 U/L), and C-reactive protein of 32.5 mg/dl (normal < 5 mg/dl). HbS was measured at 67.5%, and HbF was 1.5%. A plain chest X-ray was normal, and abdominal ultrasound showed splenomegaly (15x15x8.5 cm). A diagnosis of acute pain crisis was considered probable, so blood and urine cultures were obtained, and empirical treatment was started, including ceftriaxone, levofloxacin, low molecular weight heparin, paracetamol, and intravenous crystalloids, while waiting for transfusion.
Twelve hours post-admission and prior to the planned transfusion, the patient lapsed into a comatose state with the Glasgow Coma Scale score declining precipitously from a full 15/15 to 7/15. Neurological assessment showed centered, equally round pupils with intact responses to light. The patient required emergency intubation, and a subsequent brain computed tomography (CT) scan revealed widespread cerebral edema without signs of focal ischemia (Figure 1). Concurrent hematologic analysis identified severe hemolysis; hemoglobin levels dropped to 3.4 g/dl, while LDH and total bilirubin levels escalated to 1837 U/L and 9.0 mg/dl, respectively. The corrected reticulocyte count was measured at 1.3%. No laboratory indicators suggested ischemic injury to the kidneys, liver, or heart. Additional immunohematological evaluations detected CC ee Kell-jka antibodies and a positive result on both direct and indirect Coombs tests, leading to a diagnosis of hyperhemolysis syndrome.
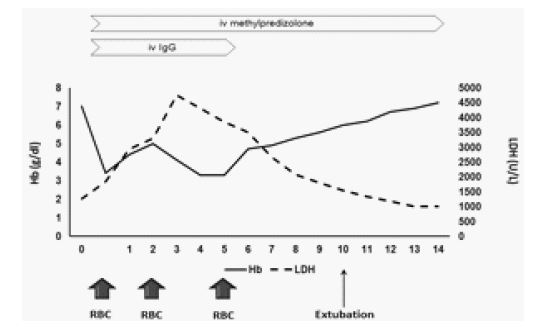
Treatment was modified with the addition of mannitol for brain edema, methylprednisolone (40 mg x 3), and gammaglobulin (400 mg/kg for 5 days) (Figure 2) for hyperhemolysis syndrome. Vancomycin was also added to the already given antibiotic therapy. The patient was transferred to the intensive care unit. A second brain CT, three days after intubation, showed remission of brain edema. While receiving the previously mentioned immunosuppressive therapy, the patient was administered a total of three units of the most compatible leukoreduced packed red blood cells on days 0, 1, and 5. Subsequently, hemoglobin levels increased, and LDH levels showed a decreasing trend starting from the sixth day after intubation (Figure 2). The patient remained intubated for 11 days. During hospitalization, repeated blood and urine cultures were constantly negative. IgM antibodies for Mycoplasma pneumoniae, Chlamydophila pneumonia, Legionella pneumophila, Parvovirus B19, Rickettsia rickettsia, typhii, and coronii were also negative. Fever resolved 12 days after admission. Methylprednisolone was gradually tapered until discontinuation after discharge.
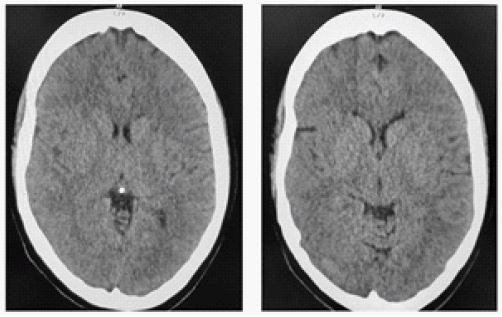
Following a sixteen-day hospitalization, the patient was discharged without neurological deficits. Recent Magnetic Resonance Imaging (MRI) did not reveal findings of acute or past ischemic stroke; however, it identified multiple hyperintense white matter lesions in T2-weighted and FLAIR sequences. These imaging findings persisted in follow-up MRIs performed in one and three months post-discharge. Despite the presence of these lesions, evaluations for demyelinating diseases were negative. One year after the episode, the patient remains free of neurological signs or symptoms. The patient was fully compliant with the recommended treatment plan, starting with a dosage of 1000 mg of hydroxycarbamide and receiving monthly infusions of crizanlizumab for eight months. The treatment was successful, as there were no further episodes of vascular occlusive crises that required hospitalization. After crizanlizumab became unavailable in Greece, the patient has solely been receiving treatment with hydroxycarbamide. To date, her response to hydroxycarbamide monotherapy remains satisfactory. Furthermore, one year after this episode, the patient has no neurological symptoms or signs.
Discussion
DHTRs are the most frequent and dangerous transfusion complications in patients with SCD. They result once a previously formed RBC alloantibody falls below detection levels, allowing transfusion of phenotypically matched but incompatible RBC units that trigger an amnestic alloantibody response [2]. This leads to accelerated destruction and hemolysis of transfused RBCs occurring days to weeks after the implicated transfusion.
SCD patients have a markedly increased risk of RBC alloimmunization compared to the general population, ranging 20-50% versus 2-3%, respectively [2]. DHTRs are similarly more common, occurring after 4.2% of non-chronic transfusions – over ten times higher than prior estimates of around 0.1% [3]. Nearly 11% of SCD patients with DHTRs died, indicating that these reactions significantly impact mortality [3].
However, DHTRs often go unrecognized or are misdiagnosed as vaso-occlusive crises, as the clinical presentation of acute anemia, hemoglobinuria, and pain mimics typical SCD complications [4]. DHTRs can also be “serologically silent” with no detectable alloantibody even when severe hemolysis occurs [5]. Routine post-transfusion antibody screening and hemoglobin A quantification improves detection, but nearly 30% of DHTRs still lack identifiable antibodies [6]. In the case of our patient, transfusions had been administered several years ago without proper documentation of the transfusion strategy employed. The patient was administered a transfusion of 3 units of blood with a limited phenotype, which posed challenges in the identification of the alloantibody.
Hyperemolysis during DHTRs presents additional risks, as hemolysis is accelerated beyond the transfused RBCs to destroy the patient’s own RBCs. Case reports describe hemoglobin levels falling as low as 1.6 g/dL during hyperemolysis, which can rapidly become fatal [7]. The exact mechanisms of hyperemolysis are unclear but may relate to complement activation by free hemoglobin and sheer RBC destruction overwhelming macrophage clearance capacity [6]. PRES is another under-recognized transfusion complication in SCD patients. It involves vasogenic edema in posterior brain regions, leading to headaches, seizures, visual disturbances and altered mental status [8]. While symptoms mimic stroke, MRI reveals reversible edema and helps differentiate PRES if obtained early [9].
PRES syndrome is a recognized complication in children with sickle cell disease (SCD), where it manifests in up to 10% of these patients, compared to only 0.4% in the general pediatric population, suggesting an increased vulnerability in SCD. Chronic anemia is thought to play a role by causing compensatory vasodilation and endothelial dysfunction. While transfusion and hypertension have been suggested as trigger factors, their links to PRES syndrome are not consistently observed [10,11]. Conversely, in adults, reports of this complication are scarce [12]. A literature review found only 12 cases of adult SCD patients with PRES syndrome [12-20] (Table 1). The majority were females (9 out of 12), aged between 19-34 (10 out of 12), with only two patients over 50. The primary clinical manifestation was seizures, with visual disturbances and headaches also being common. Hypertension was identified as a possible precipitating factor in about half of the reported cases (7 out of 12) [12,14,15,19,20]. Treatment approaches varied, but the majority of the cases resulted in the resolution of PRES symptoms.
Differentiating PRES from stroke is critical, as transfusion therapy for presumed stroke can exacerbate injury in PRES. However, diagnosis is challenging since both are presented with acute neurological changes. PRES mimicking focal strokes has also been reported [21]. Seizures occur in up to 90% of PRES cases but in only 10% of strokes, potentially aiding distinction [22]. Headache, vision changes and encephalopathy suggest PRES, while focal motor deficits or aphasia suggest stroke. Ultimately MRI with diffusion weighted imaging provides the most definitive differentiation when available [23]. Preventing DHTRs and PRES requires limiting transfusions according to evidence-based indications in SCD patients. Providing phenotype-matched RBC units based on prior alloantibody screening is also recommended [24]. For suspected DHTRs, treatment involves supportive care while avoiding further RBC transfusion due to risks of potentiating hypheremolysis. Emerging therapies like complement inhibition show promise for severe hyperhemolysis [25]. Delayed Hemolytic Transfusion Reactions (DHTR) management requires a comprehensive approach to effectively address this rare but serious complication of blood transfusions [26]. Early recognition and diagnosis of DHTR are crucial for prompt intervention. Supportive care, including close monitoring of vital signs and blood transfusion as needed, plays a vital role in managing anemia and maintaining organ perfusion. Corticosteroids, such as prednisone, have been utilized in DHTR management to suppress the immune response and reduce hemolysis. Intravenous immunoglobulin (IVIG) therapy, specifically using anti-D or anti-Kell antibodies, has also shown promising results in some cases [27]. Additionally, the use of monoclonal antibodies like rituximab and eculizumab may be considered in severe cases of DHTR associated with complement-mediated hemolysis. Rituximab works by targeting B cells involved in the immune response, while eculizumab targets complement protein C5, effectively halting the progression of hemolysis [28]. Furthermore, identifying the specific antibody causing DHTR is crucial for selecting compatible blood products in subsequent transfusions, and crossmatching should be performed to ensure compatibility and prevent further hemolysis. A comprehensive understanding of DHTR management strategies, including the use of corticosteroids, IVIG therapy, rituximab and eculizumab, is essential in providing effective treatment and ensuring patient safety.
For PRES, prompt antihypertensive therapy and eliminating triggers like transfusion are important while reversible edema persists on MRI. Anticonvulsants can help manage seizures until resolution [29]. Distinguishing PRES from stroke prevents inappropriate transfusion, though evidence guiding optimal management remains limited in SCD patients.
References
- Kato GJ, Piel FB, Reid CDM et al. Sickle cell disease. Nat Rev Dis Primers. 2018; 15:4:18010.
- Yazdanbakhsh K, Ware RΕ, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012; 120(3): 528–537.
- Narbey D, Habibi A, Chadebech P, Mekontso-Dessap A, Khellaf M, Lelievre J-D, et al. (2017). Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease. Am J Hematol. 2017; 92(12): 1340–1348.
- Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood. 2013; 122(6): 1062–1071.
- Habibi A, Mekontso-Dessap A, Guillaud C, Michel M, Razazi K, Khellaf M, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: Presentations, outcomes, and treatments of 99 referral center episodes. Am J Hematol. 2016; 91(10): 989–994.
- Balbuena-Merle R, Hendrickson JE. Red blood cell alloimmunization and delayed hemolytic transfusion reactions in patients with sickle cell disease. Transfus Clin Biol. 2019;26(2): 112-115.
- Jang JH, Yoon J, Kim JY, Jeon YW, Cho YU, Lee JW, et al. Successful management of delayed hemolytic transfusion reaction in a patient with sickle cell disease using eculizumab: A case report. J Korean Med Sci. 2015; 30(12): 1870–1873.
- Hinchey J, Chaves C, Appignani B, Breen J, Pao L, Wang A, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996; 334(8):494–500.
- Bartynski WS, Boardman JF. Distinct imaging patterns and lesion distribution in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008; 29(7): 1320–1327.
- Raj S, Overby P, Erdfarb A, Ushay HM. Posterior reversible encephalopathy syndrome: Incidence and associated factors in a pediatric critical care population. Pediatr Neurol. 2013; 49(5): 335–339.
- Khademian Z, Speller-Brown B, Nouraie SM, Minniti CP. Reversible posterior leukoencephalopathy in children with sickle cell disease. Pediatr Blood Cancer. 2009; 52(3): 373–375.
- Vargas A, Testai FD. Posterior Reversible Encephalopathy Syndrome in adult sickle-cell patients: Case series and literature review. J Clin Neurosci. 2019; 70: 249-250.
- Raj S, Killinger J, Overby P. Blood transfusion in sickle cell disease leading to posterior reversible encephalopathy syndrome (PRES). J Child Neurol. 2013; 28(10): 1284-1286.
- Parameswaran BK, Krishnan PR, Al Dossary J. Recurrent posterior reversible encephalopathy syndrome in a patient with sickle cell disease. Ann Saudi Med. 2007; 27(3): 206 –211.
- Aiyer R, Klein D, El-Sherif Y. Rare Case of Posterior Reversible Leukoencephalopathy Syndrome Secondary to Acute Chest Syndrome. Case Rep Radiol. 2016; 2016: 4346953.
- Geevasinga N, Cole C, Herkes GK, Barnett Y, Lin J, Needham M. Sickle cell disease and posterior reversible leukoencephalopathy. J Clin Neurosci. 2014; 21(8): 1329-1332.
- Landais A, Lemonne N, Julan ME. Uncommon Posterior Reversible Encephalopathy Syndrome in a Sickle-Cell Patient. J Clin Neurol. 2015; 11(3): 287–288.
- Nair A, Testai FD. Recurrent posterior reversible encephalopathy syndrome in a sickle cell patient J Natl Med Assoc. 2011; 103(2): 170-172.
- Sweany JM, Bartynski WS, Boardman JF. “Recurrent” posterior reversible encephalopathy syndrome: report of 3 cases--PRES can strike twice! J Comput Assist Tomogr. 2007; 31(1): 148-156.
- Zuccoli G, Nardone R, Rajan D, Khan AS, Cummings DD. Nonaneurysmal Subarachnoid Hemorrhage in Sickle Cell Disease: Description of a Case and a Review of the Literature. Neurologist. 2018; 23(4): 122-127.
- Terranova S, Kumar JD, Libman RB. Posterior reversible encephalopathy syndrome mimicking a left middle cerebral artery stroke. Open Neuroimag J. 2012; 6: 10-2
- Legriel S, Pico F, Azoulay E. Understanding posterior reversible encephalopathy syndrome. In Vincent JL (eds), Annual Update in Intensive Care and Emergency Medicine 2011; 1. Springer, Berlin, Heidelberg.
- Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol. 2015; 14(9): 914–925.
- Yazdanbakhsh K, Pirenne F. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood. 2018; 131(25): 2773–2781.
- Noizat-Pirenne F, Bachir D, Chadebech P, Michel M, Plonquet A, Lecron JC, et al. Rituximab for prevention of delayed hemolytic transfusion reaction in sickle cell disease. Haematologica. 2007; 92(12): 132–135.
- Noizat-Pirenne F, Bachir D, Galacteros F, Bierling P. The use of rituximab to prevent severe delayed haemolytic transfusion reaction in immunized patients with sickle cell disease. Vox Sanguinis. 2015; 108(3): 262–267.
- Thein SL, Pirenne F, Fasano RM, Habibi A, Bartolucci P, Chonat S, et al. Hemolytic transfusion reactions in sickle cell disease: underappreciated and potentially fatal. Haematologica. 2020; 105(3): 539–544.
- Vlachaki Ε, Gavriilaki Ε, Kafantari Κ, Adamidou D, Tsitsikas D, Chasapopoulou E, et al. Successful outcome of hyperhemolysis in sickle cell disease following multiple lines of treatment: the role of complement inhibition. Hemoglobin. 2018; 42(5-6): 339-341.
- Henderson JN, Noetzel M., McKinstry RC, White DA, Armstrong M, DeBaun MR. Reversible posterior leukoencephalopathy syndrome and silent cerebral infarcts are associated with severe acute chest syndrome in children with sickle cell disease. Blood. 2002; 99(2): 415–419.