
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
The pneumonia patient who refused to get better: A unique presentation of Wegener’s Granulomatosis
Shravan Gangula1*; Haripriya Bhimasani2; Sumanth Balguri3
1Coffeyville Regional Medical Center, USA.
2Deccan College of Medical Sciences, USA.
3Mercy Hospital Fortsmith, USA.
*Corresponding Author : Shravan Gangula
Coffeyville Regional Medical Center, USA.
Email: dr.shravangangula@gmail.com
Received : Jan 26, 2024
Accepted : Feb 12, 2024
Published : Feb 19, 2024
Archived : www.jcimcr.org
Copyright : © Gangula S (2024).
Abstract
Wegener’s Granulomatosis (WG) is a rare immune-mediated disorder which affects the upper respiratory tract, lungs and the kidneys. It is characterized by an inflammatory response mainly in the small vessels which leads to organ damage. Wegener’s Granulomatosis canbe challenging to diagnose on presentation alone. Radiographic studiesmay be misleading, but cultures, biopsies with immunohistochemical staining can be used to differentiate it from amyloidosis, multiple myeloma and other causes leading to renal failure. We present a case of a patient who was admitted for pneumonia who continued to get worse with antibiotics and developed renal failure. A renal biopsy revealed the diagnosis of Wegener’s Granulomatosis.
Keywords: Wegener’s Granulomatosis; Vasculitis; cANCA.
Abbreviations: WG: Wegener’s Granulomatosis; Canca: Cytoplasmic Antineutrophil Cytoplasmic Antibodies; Pr3: Proteinase 3.
Citation: Gangula S, Bhimasani H, Balguri S. The pneumonia patient who refused to get better: A unique presentation of Wegener’s Granulomatosis. J Clin Images Med Case Rep. 2024; 5(2): 2869.
Case report
A 74-year-old caucasian female with a history of controlled hypertension presented with right upper quadrant abdominal pain, nausea and vomiting of 2 days duration. The patient also reported intermittent episodes of sinusitis and shortness of breath for about a week, but no fever, cough, hemoptysis, or rash. She denied any history of bleeding problems or renal stones, although admitted to hematuria for the last few days and a single episode of epistaxis. The patient also reported a history of a 10-pound unintentional weight loss in the last month, as well as non-specific joint pains and weakness over the past few months, for which she was placed on oral diclofenac as an outpatient.
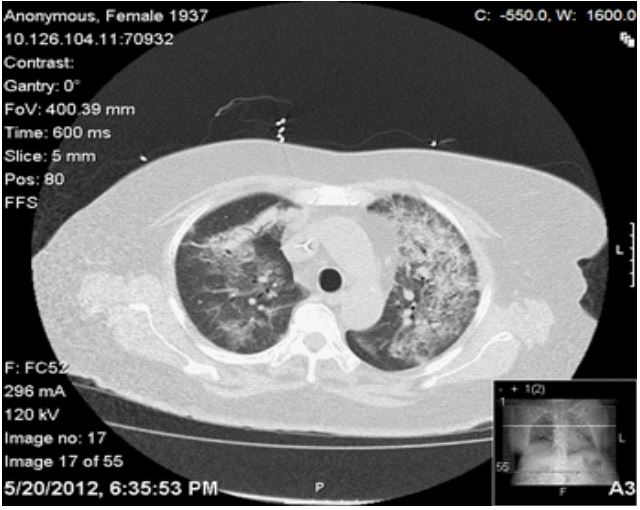
On initial exam, the patient was ill-appearing with a temperature of 98.7°F, blood pressure 138/67 mmHg, pulse 96/min and respiratory rate 22/min. A detailed physical exam was benign except for right upper quadrant tenderness, positive Murphy’s sign, and coarse breath sounds and rales bilaterally. Pertinent positives in Laboratory investigation included a leukocytosis of 11600, BUN of 34 and a creatinine of 1.92. The patient’s baseline creatinine was 0.9 about 6 months prior to admission. Urinalysis revealed 100+ RBCs, 5-10 WBCs, 1+ protein. Radiological studies confirmed that the patient had bilateral pulmonary infiltrates and a calcified gallbladder.
On admission, the patient was given intravenous moxifloxacin for the presumable pneumonia and scheduled for a laparoscopic cholecystectomy on the following day. Her elevated creatinine was felt to be a diclofenac-induced interstitial nephritis, so the medication was ceased and IV fluids were begun. Postoperatively, the patient required supplemental oxygen to maintain adequate saturations and her renal function continued to deteriorate. A repeat CT scan of the chest revealed worsening bilateral pulmonary infiltrates, although her blood and sputum cultures were negative. The moxifloxacin was changed to IV vancomycin and piperacillin- tazobactam to provide broader antimicrobial coverage. A bronchoscopy was accomplished, which found no bacteria, but suggested thrombosis.
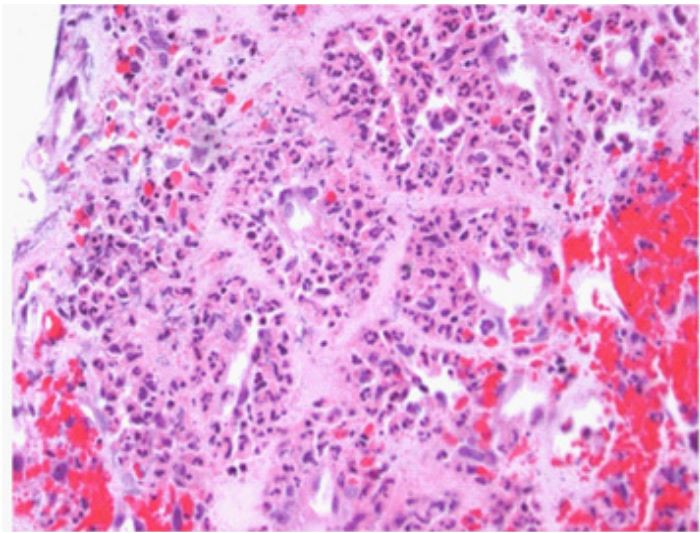
The initial diagnosis of pneumonia was no longer viable, given the bronchoscopic findings. A renal ultrasound ruled out renal artery stenosis. Immunologic studies were accomplished and revealed PR3+ ANCA. A subsequent renal biopsy to confirm the diagnosis revealed focal segmental necrotizing glomerulonephritis consistentwith atypical wegener’sgranulomatosis.
The patient was started on cyclophosphamide and mesna. Improvements in her creatinine and oxygen requirements were observed within 24 hours. As her condition continued to improve, she was ultimately discharged home under outpatient care with pulse methylprednisolone therapy
Discussion
Epidemiology: The prevalence of WG in the US is estimated to be 3 per 100,000. Overall, the Caucasian race is affected more with a male to female hospitalization ratio of 1:1. However, WG deaths are significantly more likely to occur in males [1].
Pathogenesis: The hallmark of WG includes the clinical constellation of pulmonary – renal vasculitis and serological markers – cANCA. In the initial phase, when WG is localized to the upper respiratory tract, the specificity of cANCA is about 50%. The specificity increases to 100% when WG is generalized [2]. The principal target antigen for cANCA in WG is [3]. PR3-ANCA induces invitro activation of cytokine primed Polymorphonuclear granulocytes (PMNs), resulting in degranulation of PMNs and subsequent release of reactive oxygen radicals and lytic enzymes. Activated PMNs release inflammatory mediators like tumor necrosis factor- alpha, interleukin 1 and 8 and leukotrienes which cause the pathognomic lesions of WG [4]. Genetically, WG has been associated with HLA class II and the presence of DQw7, DR4 halotypes [5]. Zycinska et al. [6] reported the association of chronic nasal staphylococcus aureus carriage with higher relapse rates in WG.
Clinical features: Unexplained constitutional symptoms are often part of the initial presentation. At presentation, 80% of WG patients do not have renal involvement. About 70% of patients present with non-specific nasal/ sinus or tracheal abnormalities which will often be attributed to upper respiratory infections or allergies. Pulmonary involvement is one of the cardinal features of WG. Cough, hemoptysis and dyspnea are the most common pulmonary symptoms. The common findings on pulmonary imaging are infiltrates and nodules [2]. Renal disease can progress from asymptomatic hematuria to fulminant glomerulonephritis in a span of days to weeks. Pulmonary symptoms coupled with the findings of hematuria and a positive C-ANCA should be further investigated with a renal biopsy [2]. Ocular manifestations have also been reported in about 28% of the patients with WG with visual loss being reported in 8% of the cases affected [7]. WG can manifest as conjunctivitis, dacryocystitis, scleritis, proptosis and retinal damage in decreasing order of frequency.
Treatment: The treatment of WG can be divided in to initial immunosuppressive therapy and maintenance immunosuppressive therapy. Induction of complete remission is the goal of immunosuppressive therapy and remission is defined as the absence of active disease. Initial immunosuppressive therapy typically consists of cyclophosphamide and glucocorticoids. Cyclophosphamide is given orally in a dose of 1.5 to 2 mg/kg per day and is continued until remission is induced. Leukopenia can be a life threatening complication of cyclophosphamide therapy, and white blood cell count should be regularly monitored while on cyclophosphamide [8]. Glucocorticoid therapy can be in the form of pulse methylprednisolone therapy (7-15 mg/kg) or oral prednisone therapy (1 mg/kg). The initial dose is continued for 2-4 weeks till remission is achieved and then slowly tapered with agoal of reaching 20 mg/kg by the end of 2 months [9].
Rituximab can be an effective alternative to cyclophosphamide for the initial treatment of patients with WG [10]. Low dose weekly oral methotrexate can also be used as initial therapy if there is no organ or life threatening disease [9]. Plasma exchange can be used to treat patients with pulmonary hemorrhage [11]. The preferred drugs for maintenance therapy in patients with remission are methotrexate and azathioprine. Weekly oral methotrexate started within one to two days of the last cyclophosphamide dose is started at a dose of 0.3 mg/ kg per week and increased in 2.5 mg increments every week if tolerated to a dose of 20 to 25 mg per week. If remission is sustained for 2 years or longer, then methotrexate can be tapered by 2.5 mg each month until discontinuation [12]. Azathioprine used at 2 mg/kg per day, then reduced to 1.5 and 1.0 mg/kg per day after 12 and 18 months respectively has equivalent efficacy in maintaining remission when compared to methotrexate [13]. Low dose corticosteroid therapy is initially continued in most patients receiving maintenance therapy. Other drugs like mycophenolate, rituximab and trimethoprim-sulfamethoxazole can also be used for maintenance therapy in the patients who do not tolerate azathioprine or methotrexate.
Conclusion
Wegener’s Granulomatosis is a rare but potentially lethal condition if not treated. It needs to be considered in the differential diagnosis of a pneumonia patient with atypical features who do not get better with traditional treatment strategies.
References
- Cotch MF, Hoffman GS, Yerg DE, et al. The epidemiology of Wegener’s granulomatosis. Arthritis Rheum. 1996; 39: 87-92.
- Hoffman GS. Wegener’s granulomatosis. Curr Opin Rheum. 1993; 5(1): 11-17.
- Jenne DE, Tschopp J, Lodemann J, et al. Wegener’s autoantigen decoded. Nature 1990; 346: 520.
- Falk RJ, Terrel RS, Charles LA, et al. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990; 87: 4115-19.
- Spencer SJ, Burns A, Gaskin G, et al. HLA class II specificities in vasculitis with antibodies to neutrophil cytoplasmic antigens. Kidney Int. 1992; 41(4): 1059-1063.
- Zycinska K, Wardyn KA, Zeilonka TM, et al. Chronic crusting, nasal carriage of staphylococcus aureus and relapse rate in pulmonary wegener’s granulomatosis. J physiol pharmacol. 2008; 59 suppl 6: 825-831.
- Bullen CL, Leisegang TJ, McDonald TJ, et al. Ocular complications of Wegener’s granulomatosis. Ophthalmology. 1983; 90: 279-90.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener’s granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992; 116: 488.
- Langford CA, Talar-Williams C, Sneller MC. Use of methotrexate and glucocorticoids in the treatment of Wegener’s granulomatosis. Long termrenal outcomein patients with glomerulonephritis. Arthritis Rheum. 2000; 43: 1836.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA- associated renal vasculitis. N Engl J Med. 2010; 363: 221.
- Klemmer PJ, Chalermskulrat W, Reif MS, et al. Plasmapheresis therapy for diffuse alveolar hemorrhage in patients with small vessel vasculitis. Am J Kidney Dis. 2003; 42: 1149.
- Langford CA, Talar-Williams C, Sneller MC, et al. Use of cyclophosphamide – induction methotrexate – maintenance regimen for the treatment of Wegener’s granulomatosis: extended follow-up and rate of relapse. Am J Med. 2003; 114: 463.
- Pagnoux C, Mahr A, Hamidou MA, et al. Azathioprine or methotrexate maintenance for ANCA- associated vasculitis. N Engl J Med. 2008; 359: 2790.