
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Research Article - Open Access, Volume 5
Identification of one novel homozygous nonsense mutations in DYSF in two siblings with muscular dystrophy product of consanguineous marriage from Iran
Sara Samanian1,2; Raheleh Torabi1 ; Mina Mohammadi Sarband4 ; Fakhimeh Hasemnia1 ; Banafshe Afkari1 ; Frouzandeh Mahjoubi1,3*
1Solale Medical Genetic Laboratory, Genetic Foundation of Tehran, Tehran, Iran.
2Department of Anatomy, Center of Anatomy and Cell Biology, Medical University of Vienna, Vienna, Austria.
3Medical Biotechnology Department, National Institute of Genetics Engineering and Biotechnology (NIGEB), Tehran, Iran.
4Ameneh Genetic Counseling, Shemiranat Welfare Center, State Welfare Organization, Tehran, Iran.
*Corresponding Author : Frouzandeh Mahjoubi
Founder members of Genetic Foundation of Tehran,
Medical Biotechnology Department, National
Institute of Genetics Engineering and Biotechnology
(NIGEB), Tehran, Iran.
Email: frouz@nigeb.ac.ir
Received : Feb 04, 2024
Accepted : Feb 20, 2024
Published : Feb 27, 2024
Archived : www.jcimcr.org
Copyright : © Mahjoubi F (2024).
Abstract
Mutations in DYSF are associated with autosomal recessive muscular dystrophy, a major public health issues with estimated global prevalence of 3.6 per 100,000 individuals. Two suspected siblings for muscular dystrophy were referred for clinical and genetic evaluation. Clinical evaluation, and Whole-Exome Sequencing (WES) were used to characterize etiology in one of two affected siblings from product of consanguineous marriage of Iranian descent. Sanger sequencing were applied to confirm the genotype of other sibling and parents for the found mutation in WES. Whole-exome sequencing showed deletion of 21 bases (c.3448_3468del) in exon 32 of Dysferlin (DYSF) gene (NM_003494.1) in one of siblings. Sanger sequencing for this mutation in DYSF (Exon 32) confirmed homozygosity in other affected sibling and heterozygosity in their healthy parents and healthy sibling. The one known variant found in DYSF which lead to muscular dystrophy as clinical manifestation. Investigation effect of variant on protein structure revealed that not only this deletion caused truncated protein, but also decreased the beta structures in product protein which may disrupt function of its.
Keywords: Dysferlin; Muscular dystrophy; Iran; Deletion.
Citation: Samanian S, Torabi R, Sarband MM, Hasemnia F, Mahjoubi F, et al. Identification of one novel homozygous nonsense mutations in DYSF in two siblings with muscular dystrophy product of consanguineous marriage from Iran. J Clin Images Med Case Rep. 2024; 5(2): 2883.
Introduction
Muscular dystrophy is a group of inherited neuromuscular conditions that cause progressive weakness and loss of muscle mass. Necrotic and regenerating fibers, an increase in fiber size variation, fiber splitting and centrally located myonuclei are characteristics of muscular dystrophies. There are many kinds of muscular dystrophy with specific age of onset, rate of progression, and pattern of inheritance and classified by their clinical presentation, the distribution and extent of muscle weakness which show specific signs and symptoms in different muscles. In most common variety, the symptoms begin in childhood, commonly in male patients. Other types don’t surface until adulthood [1]. In muscular dystrophy, the necrosis and replacement of muscle with fatty and fibrous tissue happens due to rounds of degeneration and regeneration of muscle fibers. Fragile dystrophic skeletal muscle plasma membranes caused by muscle contraction leads to muscle cell degeneration [2]. More than 30 genes have been linked to muscular dystrophies by cutting-edge molecular genetic techniques like positional cloning and candidate gene analysis [1]. Homo sapiens Dysferlin gene (DYSF; OMIM# 603009) with cytogenetic location of 2p13.2 and encoding 2,080 amino acids plays an important role in muscle fiber repair [3]. Pathogenic mutations and putative phenotypeinfluencing genetic variations in DYSF gene could be responsible for muscular dystrophies in three different phenotypes including Miyoshi myopathy, limb-girdle muscular dystrophy type 2B, and distal anterior compartment myopathy which identified by novel next-generation sequencing technologies such as WholeExome Sequencing (WES) [4,6]. Patients with homozygous or compound heterozygous mutation in DYSF gene in previous studies displayed muscular dystrophy [7]. We reported novel 21 bases deletion in DYSF exon 32 in two siblings with progressive muscular dystrophy/Limb-girdle, type 2B (OMIM: 253601) which could confirm the importance of this exon in DYSF function.
Materials and methods
Patient data: In this study, we reported two siblings (Patients I and II), product of consanguineous marriage of healthy parents (Individuals IV and V) in Iran. Patient I was asymptomatic until he reached age 19 years. At this age he noticed gait problem and giving away particularly in left side. Clinical examination found weakness of both legs, restricted of angle plantar, and dorsiflexion. Absent of ankle reflex and hypoactive knee jerks, was also detected. CPK level was 8000 (mcg/L). Myopathy EMG finding mainly in distal muscles of lower and upper limbs in diagnosis of myopathy in process was reported. Immunochemical study of sarcolemma proteins, was all in favor of dysferlinopaty. His affected sister (Patient II) has also gone through the same process. They also have an apparently healthy sister (Patient III).
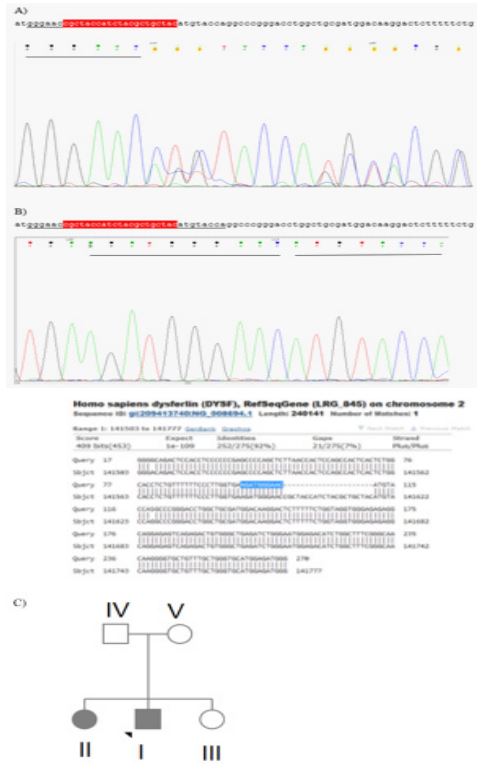
Experimental methods: Appropriate written informed consent for genomic analysis and permission to use the samples and data for research purpose was obtained from the patients. DNA was extracted from peripheral blood leukocytes of three patients using a commercial kit (High Pure PCR Template Preparation, Roche). To determine the molecular etiology, WholeExome Sequencing (WES) was performed on peripheral blood karyotype of one of siblings. Whole-exome sequencing on DNAs was enriched for exonic regions with SureSelect 38 Mbp. All exon kit v. 7.0 (Agilent Technologies) was prepared according to manufacturer protocols, and 75×2 bp paired-end sequenced on HiSeq 2000 (Illumina Inc.), with a 80-120× mean coverage. The sequencing quality was confirmed using FastQC 11.5 software (Bittencourt a. 2010). Variants were filtered in the preliminary whole exome data analysis was performed through Burrows– Wheeler Aligner (BWA; Li & Durbin, 2009) and the Genome Analysis Toolkit (GATK) software (McKenna et al. 2010) to generate a Binary Alignment Map (BAM) and a Variant Call Format (VCF) file, respectively. Annotations of the VCF files were carried out through the wANNOVAR software (Wang et al. 2010), and the data were manually analyzed for the presence of candidate pathogenic variants. Variants were filtered out in different human population databases. The local NGS database (www.iranome.ir), as well as public databases (the 1000 Genomes Project [Consortium, 2015]), the Genome Aggregation Database (gnomAD), the Genome Aggregation Consortium (ExAC; Lek et al. 2016), ESP6500 (Fu et al. 2013), and dbSNP 137 were also investigated. Pathogenicity of the variants were assayed using prediction methods (PolyPhen-2 (Adzhubei et al. 2013), SIFT (Ng & Henikoff, 2003), MutationTaster (Schwarz et al. 2014), PROVEAN (Choi & Chan, 2015), and in silico nucleotide conservation from Genomic Evolutionary Rate Profiling (GERP) scores (Pollard et al. 2010)). VarSome database, a search engine for human genomic variation, was also used for classifying candidate variants according to the criteria set by the American College of Medical Genetics (ACMG; Richards et al. 2015). The identified mutations were validated using Sanger sequencing. The online version of Primer 3 software was used to design primers flanking candidate variants. The forward and reverse primers for c.3448_3468del are as follows, respectively: F (exon 32) _ tgtggtttccctcattcagc, r_ tgacagcacccatctccat. Regions were amplified and sequenced using ABI 3500 Genetic Analyzer (Applied Biosystems Inc., 850 Lincoln Center Drive). Segregation analysis using Sanger sequencing was performed for two other siblings and their parents in at the position of mutation.
Structure analysis: The Dysferline structural information including domains and their amino acid sequences were extracted from UNIPORT (Entry Key: O75923: https://www.uniprot. org). The predicted features of wild and mutant structures were obtained by PredictProtein (https://predictprotein.org/) and revealed in Figure 1 containing solvent accessibility, secondary structure, and conservation. The tertiary structures of C2_4 domain and mutant C2_4 domain were predicted by Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2).
Results
In the son of Family (Patient I), a homozygous missense mutation DYSF(NM_003494.4): c.3448_3468del in exon 32 of DYSF (NM_003494.4; Figure 1) was identified by WES. The variants are not present in population databases Iranom. Detailed computational analysis of p.Asn1150_Cys1156del mutation by various prediction methods (PolyPhen-2, SIFT, MutationTaster, and PROVEAN) revealed mutation as Variant of Uncertain Significance (VUS) according to the ACMG criteria. This mutation cause deletion of amino acids of Asparagine 1150 to Cysteine 1156 (p.Asn1150_Cys1156del). Sanger sequencing confirmed homozygosity of his affected sister and heterozygosity of their healthy sister (Patient III) and also their parents (Patient IV and V) (Figure 2). The predicted features of wild and mutant structures were obtained by PredictProtein (https://predictprotein. org/) and revealed in Figure 1 containing solvent accessibility, secondary structure, and conservation. Based on these results the beta strand decreased after deletion. The peptide will be exposed in all regions. We also analyzed and compared predicted secondary and third structure of wild type and mutant C2 domain product proteins which confirmed significant decrease in beta strand in mutant one.
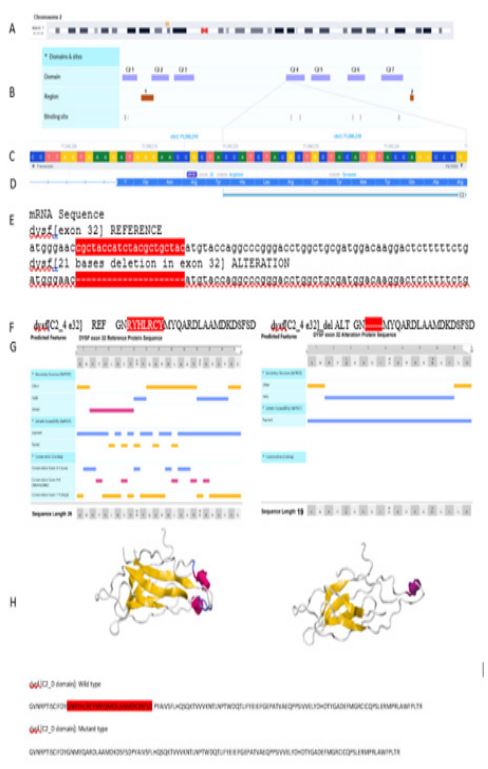
Table 1: The previous reported pathogenic mutations in DYSF exon 32 in dbSNP Short Genetic Variations (https://www.ncbi.nlm. nih.gov/snp/).
| Name | dbSNP ID |
|---|---|
|
NM_001130987.2(DYSF):c.3498T>A (p.Tyr1166Ter) GRCh37Location: 71817342 |
rs758944159[11] |
|
NM_001130987.2(DYSF):c.3498_3499delinsAA (p.Tyr1166_Gly1167delinsTer) 71817342 – 71817343 |
rs398123781 |
|
NM_001130987.2(DYSF):c.3517del
(p.Arg1173fs) 71817361 |
rs2093220811 |
|
NM_001130987.2(DYSF):c.3532C>T (p.Gln1178Ter) 71817376 |
rs886042091 |
|
NM_001130987.2(DYSF):c.3540_3541del (p.Asp1181fs) 71817383 - 71817384 |
not provided |
|
NM_003494.3(DYSF):c.3504dup
(p.Lys1169Glnfs) 71817401 - 71817402 |
rs886042504 |
|
NM_001130987.2(DYSF):c.3559A>T (p.Lys1187Ter) 71817403 |
rs2093222376 |
|
NM_001130987.2(DYSF):c.3566_3567del (p.Ser1189fs) 71817408 - 71817409 |
rs886042827 |
|
NM_001130987.2(DYSF):c.3571dup (p.Ser1191fs) 71817410 - 71817411 |
rs766341386 |
|
NM_003494.3(DYSF):c.3516_3517delTT 71817411 - 71817412 |
rs766341386 |
|
NM_001130987.2(DYSF):c.3574+1G>A 71817419 |
not provided |
Discussion
A group of inherited neuromuscular conditions that cause progressive weakness and loss of muscle mass called muscular dystrophy are characterized by necrotic and regenerating fibers, an increase in fiber size variation, fiber splitting and centrally located myonuclei. DYSF gene variations have been considered as critical gene which responsible for Muscular dystrophy with various symptoms with different age of onset [8,9]. Exon 32 of DYSF has been shown to be dispensable for dysferlin functions [10]. However, there are some reported cases with mutations in DYSF exon 32 which showed clinical manifestations of muscular dystrophy (Table 1). In a mutation analyses of DYSF in 14 Italian patients from 10 unrelated families with a deficiency of dysferlin protein, two siblings with same mutation were reported clinical features were different. The brother showed proximal dominant muscle involvement, while the sister showed distal myopathy. It seems that other genetic or epigenetic factors can impact on phenotype of patients with same mutation [11]. In a rare report, a female patient with homozygous deletion of 21 nucleotides within exon 32 (NM_003494.4: c.3451_3471del: p.Arg1151_Tyr1157del). In the age of 20, she showed first symptom as pain and then gradually proximal-distal weakness in the muscles which followed by partial biceps atrophy and Calf atrophy. Her Creatine kinase serum level was 9000. The initial diagnosis for her was Polymyositis which eventually diagnose as Dysferlinopathy [12]. This could confirm that, in spite of previous studies in frame deletion in DYSF exon 32 could show clinical manifestations of Muscular dystrophy in youth. In a genomic analysis of the DYSF coding sequence among 34 unrelated patients from various ethnic origins, one patient with the diagnosis of dysferlinopathy showed a mild reduction of dysferlin expression on Western-blot along with a significant increase of Creatine Kinases in the serum. However, he remained asymptomatic at age 11. He was heterozygout for a nonsense mutation (c.3477C>A: p.Tyr1159Ter) in C2_4 domain of Dysf [13]. In this report, we describe two Iranian siblings, born from consanguineous marriage of Persian descent, with symptoms of muscular dystrophy in youth whose WES analysis revealed homozygous deletion of 21 bases (c.3448_3468del) in DYSF gene exon 32 in male sibling which confirmed in him and his female sibling by Sanger sequencing. The heterozygosity of their parents were also showed by same method. This mutation leads to deletion of seven amino acids from position 1150 to 1156. It seems that this in frame deletion in this exon cause structural and conformational changes in produced protein. Dysferlin is known to contain various functional domains, including seven different C2 domains [1,7], three Fer domains (FerI, FerA, and FerB), and two DysF domains, for which limited information is available on their function and essentiality [14]. The N-terminal portion of the C2D domain of Dysferlin is encoded by exons 32, 33 and 34 whose deletion would maintain the open reading frame of Dysferlin mRNA would most likely lead to loss of function of the entire domain. DYSF exon 32 is a small exon (78 bps) which encodes the fourth domain of C2 [10]. Studies have shown that severely damaged exon 32 can lead to the production of a dysferlin without this exon but highly functional. Therefore, it was suspected that the fourth C2 domain is (at least partially) redundant [15]. Howevr, the analysis of the our found variant showed that protein product has shorter beta sheet structure in region of this deletion which lead to misfolding, aggregation within the endoplasmic reticulum, and amyloidogenesis. Thus, we assumed that this 21 base deletion in mRNA was translated into a protein with an internal truncation of dysferlin’s C2_4 domain with lower stability and disrupted function. The predicted features of mutant peptide in comparison with wild type revealed significant decrease in beta strand in secondary structure which could disrupt the function. The results showed more exposed regions in mutant protein structure which may increase the sensitivity of its structure which lead to instability. The comparison of C2_4 domain product protein (including exon 32) with mutant one by predicting pdb models confirmed significant decrease in percentage of beta structure. This could suggest that exon 32 of DYSF gene could be important in structure and function of C2 domain of DYSF.
Conclusion
In conclusion, we report two patients diagnosed with muscular dystrophy carrying a known homozygous deletion in frame in DYSF. Although exon 32 previously was known as uncritical exon in Dysf product protein function, we reported a 21 deletion in frame of exon 32 which lead to muscular dystrophy in mutant patients.
Acknowledgments: The authors have no conflict of interest to declare. We would like to thank the patients’ family for their cooperation in this study.
References
- Kanagawa M and T Toda, The genetic and molecular basis of muscular dystrophy: roles of cell–matrix linkage in the pathogenesis. Journal of Human Genetics. 2006; 51(11): 915-926.
- Dubuisson N. et al. Histological Methods to Assess Skeletal Muscle Degeneration and Regeneration in Duchenne Muscular Dystrophy. Int J Mol Sci. 2022; 23(24).
- Mojbafan M. et al. Genetic variability in Iranian limb-girdle muscular dystrophy type 2B patients: An evidence of a founder effect. Mol Genet Genomic Med. 2019; 7(12): 1029.
- Potulska-Chromik A. et al. Pathogenic Mutations and Putative Phenotype-Affecting Variants in Polish Myofibrillar Myopathy Patients. J Clin Med. 2021; 10(5).
- Cacciottolo M. et al. Muscular dystrophy with marked Dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur J Hum Genet. 2011; 19(9): 974-80.
- Spadafora P. et al. A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B. International Journal of Molecular Sciences. 2022; 23: DOI: 10.3390/ijms23168932.
- Bushby KM. Dysferlin and muscular dystrophy. Acta Neurol Belg. 2000; 100(3): 142-5.
- Charnay T. et al. Retrospective analysis and reclassification of DYSF variants in a large French series of dysferlinopathy patients. Genetics in Medicine. 2021; 23(8): 1574-1577.
- Wang N. et al. The clinical, myopathological, and molecular characteristics of 26 Chinese patients with dysferlinopathy: a high proportion of misdiagnosis and novel variants. BMC Neurology. 2022; 22(1): 398.
- Barthélémy F. et al. Dysferlin Exon 32 Skipping in Patient Cells. Methods Mol Biol. 2018; 1828: 489-496.
- Kawabe K. et al. Dysferlin mutation analysis in a group of Italian patients with limb-girdle muscular dystrophy and Miyoshi myopathy. Eur J Neurol. 2004; 11(10): 657-61.
- Fatehi F. et al. Dysferlinopathy in Iran: Clinical and genetic report. J Neurol Sci, 2015. 359(1-2): 256-9.
- Nguyen, K., et al., Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat.2005; 26(2): 165.
- Muriel J. et al. The C2 domains of dysferlin: roles in membrane localization, Ca(2+) signalling and sarcolemmal repair. J Physiol. 2022; 600(8): 1953-1968.
- Wein N. et al. Efficient bypass of mutations in dysferlin deficient patient cells by antisense-induced exon skipping. Human Mutation. 2010; 31(2): 136-142.