
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Pyoderma gangrenosum revealing Behçet’s disease: A case report and a review of the literature
Bachir Cherif A1*; Bennouar S2; Bellahmer S1; Djebbar Y1
1Department of Internal Medicine, Faculty of Medicine, University Blida, Algeria.
2Laboratory of Biochemistry, Faculty of Medicine, University Blida, Algeria.
*Corresponding Author : Bachir Cherif A
Department of Internal Medicine, Faculty of Medicine, University Blida, Algeria.
Email: ghani11bc@yahoo.fr
Received : Feb 06, 2024
Accepted : Feb 22, 2024
Published : Feb 29, 2024
Archived : www.jcimcr.org
Copyright : © Cherif BA (2024).
Abstract
Pyoderma Gangrenosum (PG) is a rare condition that clinically manifests itself as one or more painful ulcerations with an inflammatory border and purulent hobbles. They spread rapidly, and the chronic course is sometimes rapidly fatal. The association between PG and Behçet’s Disease (BD) is rare. Only 16 cases of PG associated with BD have been reported in the literature. In the majority of these cases, PG is diagnosed after the associated disease. Nevertheless, it may precede the onset, or be the telltale sign of an underlying disease. We present the case of a patient with the association of both conditions. The diagnosis was made early, and the course was favorable after the start of treatment, with net regression of symptoms after 12 weeks, contrary to what has been described in the literature.
Keywords: Pyoderma gangrenosum; Behcet’s disease; Aphtosis; Ulceration; Pathergy test; Associated disease.
Citation: Cherif BA, Bennouar S, Bellahmer S, Djebbar Y. Pyoderma gangrenosum revealing Behçet’s disease: A case report and a review of the literature. J Clin Images Med Case Rep. 2024; 5(2): 2888.
Introduction
Pyoderma Gangrenosum (PG) is a scarce clinical condition characterized by painful ulceration(s) with an inflammatory border and a purulent hutch. These ulcerations spread rapidly and evolve chronically. PG is currently classified as a neutrophilic dermatosis [1]. The first description was made by Louis Brocq in 1908 under the term “geometric phagedenism”. This term has been proposed to describe ulcerations with a pronounced trend to extend both superficially and deeply [1]. In 1930, Brunsting, Goeckermann and O’Leary adopted the current name. They described necrotic, extensive, painful ulcerations with bluish, undermined margins, surrounded by an erythematous areola in 5 patients, 4 of whom had ulcerative colitis [2]. They thought that this pathology was linked to an infectious condition. Further studies have broadened the clinical concept of PG and identified other related diseases. In most cases, PG is associated with chronic inflammatory bowel disease, rheumatic disease, haemopathy or neoplasia. The term “pyoderma” had already been used to describe “a purulent skin infection caused by pyogenic germs”. The term “gangrenosum” was added to emphasize the ulcerative and necrotizing nature of this dermatosis [3]. However, this term ought to have been considered inappropriate, as it was early demonstrated that PG is neither infectious nor gangrenous. In upto 50% of cases, PG is associated with inflammatory bowel disease such as Ulcero-Hemorrhagic Recto-Colitis or Crohn’s disease [4], polyarthritis (either seronegative or rheumatoid), hematological malignancies including acute and chronic myeloid leukemia, myelodysplastic syndrome, or monoclonal gammopathy (especially IgA) with or without lymphoma [5]. Less frequently, other associations have been described, such as lupus erythematosus, Takayasu’s disease, sarcoidosis or Behçet’s disease [6].
Observation
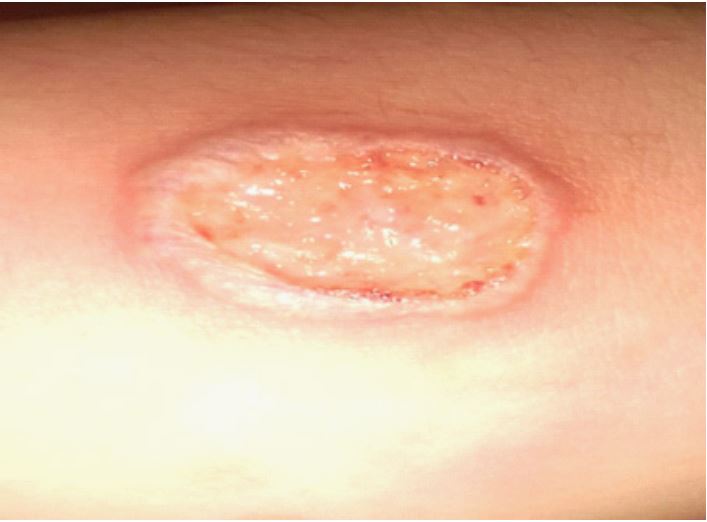
Here is a case report of a patient BN, aged 45 years, from and living in Laghouat (a city located in the northern Algerian Sahara, 329 km from the capital Algiers), who presented since 2018 recurrent buccal aphthae (>03 episodes/year) resistant to local treatment. She attended our department of internal medicine à Blida (45 km from the capital Algiers) with a round, erythematous-edged ulceration with a purulent background (Figure 1). which had progressively spread to both upper limbs and had been evolving for several months. Bacteriological samples were all negative.
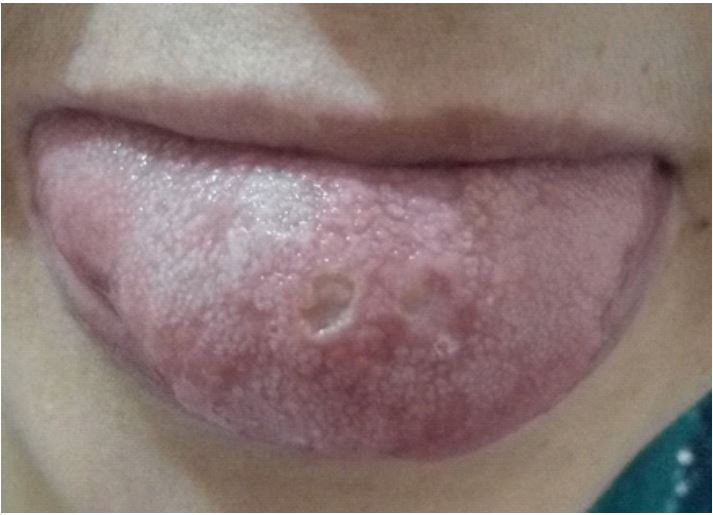
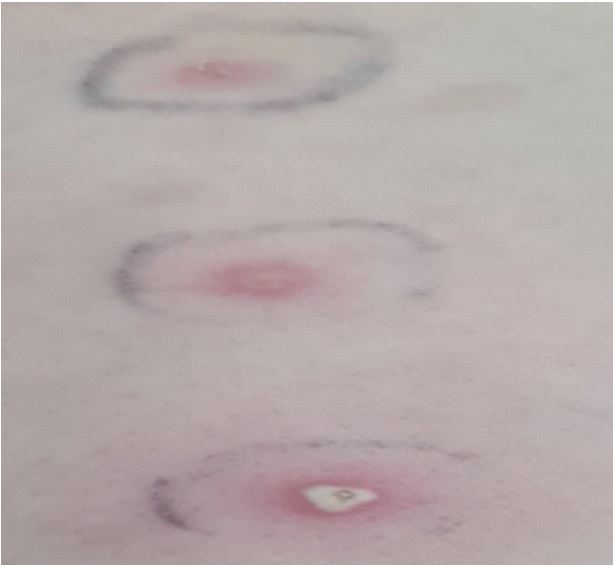
The anatomopathological assessment of a skin biopsy from the edge of a lesion showed a polymorphic dermal infiltrate with a predominance of neutrophils. The infectious test was negative, however, and given the patient’s geographical origin from the high plateaux, a high-risk area for cutaneous leishmaniasis, she was put on Glucantime as a trial treatment for 03 months, but no improvement was noted. Clinical examination upon admission revealed oral aphthae (Figure 2) with ulcerations lined with a whitish coating, genital aphthae with indelible scarring, pathergy test was performed and found positive (Figure 3), therefore according to the ACR/EULAR 2013 criteria our patient has 6 points/4 to retain the diagnosis of Behçet’s disease (BD).
The patient was subsequently treated with colchicine 1 mg/ day, corticotherapy 0.7 mg/kg/d and an immunosuppressant of the azathioprine type at a dose of 2 mg/kg/d.
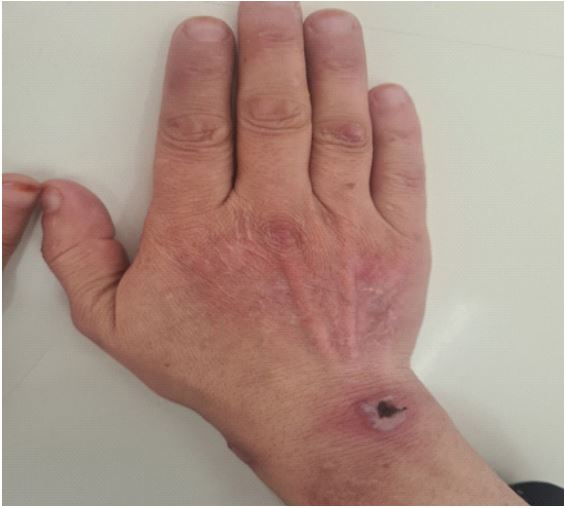
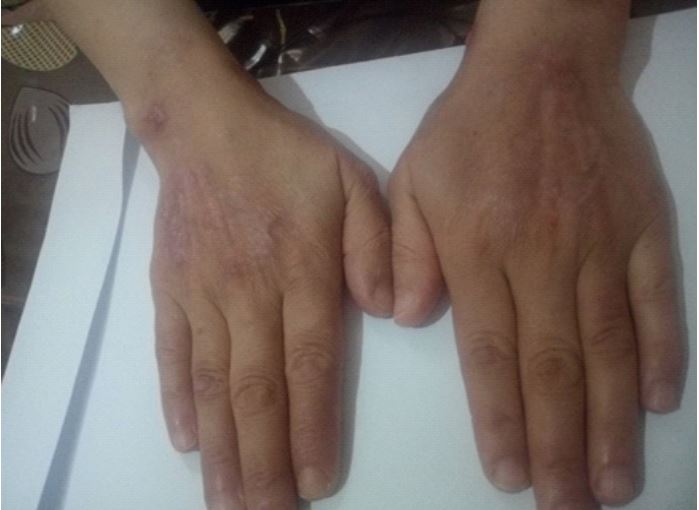
Evolution was favorable with the initiation of this therapeutic approach, marked by a significant regression of cutaneous symptoms (skin healing) within 12 weeks (Figures 4 and 5).
Discussion
The originality of our observation lies in the rare association of PG with BD. Our patient was diagnosed with BD according to the criteria of the International study group for Behcet’s disease [7]. The diagnosis of Behçet’s disease is retained if the total of symptoms is ≥4 points. Our case had 6 out of 4 points. She had buccal aphthae (two points), genital aphthae with indelible scarring (two points), skin involvement (one point) and a positive pathergy test (one point).
PG is a rare, chronic neutrophilic dermatosis that affects mainly adults and can take four anatomical and clinical forms: Ulcerative, bullous, pustular and vegetative. The diagnosis of ulcerative PG in our patient was based on the evocative clinical aspect and the absence of any other infectious or vascular cause. Ulcerative PG, the classic form, consists of a large, very painful, centrifugally expanding ulceration with undermined, purplish margins and a purulent background. It mainly affects adults aged between 25 and 55 years [8].
In our case, the cutaneous lesions were located on the two upper limbs, whereas according to the literature, PG occurs predominantly on the lower limbs (75-80%) and the upper limbs (77%) in atypical forms [9]. Most often, PG appears on the lower limbs, with a preference for the pretibial areas, but it can affect the entire mucocutaneous surface. Numerous other localizations have been reported: the thorax, hands, trunk, head, neck, around a stoma, upper aerodigestive tract, eyes, and genital mucosa [10]. PG can mimic inflammatory disease [11]. Around 50% of patients with PG have an associated disease. First and foremost are chronic inflammatory bowel diseases: ulcerative colitis and Crohn’s disease are each found in 10-15% of cases. However, during the course of Crohn’s disease or ulcerative colitis, less than 3% of patients develop PG [10]. In around 30% of cases, PG is associated with a rheumatic condition, mainly rheumatoid arthritis. In this case, it generally has a poorer prognosis than other PGs: in a study conducted over 2 years, 23.4% of PGs associated with arthritis were cured, compared with 78.9% for other PGs [12]. There is one particular rheumatic condition that is always associated with PG: the PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum and acne) [13].
The association between PG and BD is uncommon. Since Munro and Cox’s first case in 1988 [14], only 16 cases of PG associated with BD have been reported in the literature [14-18]. In the majority of these cases, PG is diagnosed following the associated disease. Although, it may precede the onset of, or be the telltale sign of, an underlying disease [19]. In the majority of cases, this was an ulcerative form of PG, as in our case. In 70% of cases, ulcerative PG is associated with systemic disease [20]. The bullous form was reported in one case and the vegetative form in two cases [8]. These patients had a predominance of digestive involvement during the course of BD, unlike our patient who had a cutaneous-mucosal and articular form without digestive involvement [21,23]. Indeed, this may be an underdiagnosed association, since the differential diagnosis between a cutaneous ulceration in Behçet’s disease and a PG may be challenging even after histopathological assessment [24]. It is often difficult to distinguish between the clinical manifestations of BD and PG in the same patient. Both BD and PG belong to the category of neutrophilic dermatoses in which primary pustular lesions appear, and neither has a characteristic histological presentation [25].
There is no consistently effective, well-codified treatment for PG; it should be individualized for each patient, depending on the severity of the PG, potential associated systemic disease and the toxicity of the molecules used.
Treatment of PG is generally based on systemic corticosteroid therapy alone or in combination with Thalidomide or Dapsone [26]. In cases of corticoresistant PG, the use of other immunosuppressive agents, such as tacrolimus, cyclosporine, mycophenolate mofetil and azathioprine, may be effective. At present, anti-TNFa is a promising treatment for refractory PG [26]. These therapeutic agents are used to treat both PG and BD [27]. Consequently, management should take both pathologies into account, and focus on corticosteroids and immunosuppressants such as cyclosporine, azathioprine or cyclophosphamide.
The clinical evolution of our patient under treatment was very favourable, with rapid regression of cutaneous lesions. The prognosis of PG depends on the associated disease and the severity of the clinical form. In the majority of published cases, PG regressed after a short treatment duration [28]. Some never develop PG again, while others may experience new onset episodes. Some PGs have a chronic or recurrent course, leading to prolonged treatment. Recurrence was estimated at 70% in patients treated with prednisolone and 66% in those treated with cyclosporine [29].
Conclusions
Behçet’s Disease (BD) is a multisystemic vasculitis with a characteristic triad of oral-genital bipolar aphthosis and uveitis. Pyoderma Gangrenosum (PG) is a neutrophilic dermatosis rarely associated with BD, and this case highlights the relevance of considering all associations with BD, even the least common, such as PG. Early intervention is crucial, but remains difficult, with a sometimes fatal prognosis.
Conflicts of interest: None.
References
- Krischer J, Modiano P, Saurat J-H. Pyoderma gangrenosum. In Dermatologie et infections sexuellement transmissibles, 5e éd. Masson, Paris. 2009; 555-7.
- Brunsting LA, Goeckerman WH, O’Leary IA. Pyoderma gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1930; 22: 655-80.
- Fahri D, Wallach D, Avril MF. Le pyoderma gangrenosum a 100 ans. De Louis Brocq aux biothérapies. Rev Prat. 2008; 58: 457-61.
- Graham JA, Hansen KK, Rabinowitz LG, et al. Pyoderma gangrenosum in infants and children. Pediatr Dermatol. 1994; 11: 10-7.
- Carron PN, Yerly S, Ksontini R, Calandra T, Meylan P. Pyoderma gan grenosum: défi diagnostique et thérapeutique. Rev Med Suisse. 2008; 4: 1938-43.
- ingh G, Sethi A, Okade R, et al. Bullous pyoderma gangrenosum: a presentation of childhood Behcet’s disease. Int J Dermatol. 2005; 44: 257-8.
- The International Criteria for Behçet’s Disease (ICBD) The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Euro Acad Dermatol Venereol. 2014; 28(3): 338-347.
- Crowson AN, Mihm MC Jr, Magro C. Pyoderma gangrenosum: a review. J Cutan Pathol. 2003; 30: 97-107.
- Benett ML, Jackson JM, Jorizzo JL, Fleischer AB Jr, et al. Pyoderma gangrenosum. A comparison of typical and atypical forms with emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000; 79: 37-41.
- Wollina U. Pyoderma gangrenosum – a review. Orphanet J Rare Dis. 2007; 2: 19.
- Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002; 347: 1412-8.
- Charles CA, Bialy TL, Falabella AF, Eaglstein WH, Kerdel FA, Kirsner RS. Poor prognosis of arthritis-associated pyoderma gangrenosum. Arch Dermatol. 2004; 140: 861-4.
- Brenner M, et al. Targeted treatment of pyoderma gangrenosum in PAPA syndrome with recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009; 161: 1199 -201.
- Munro CS, Cox NH. Pyoderma gangrenosum associated with Behçet’s syndrome--response to thalidomide. Clin Exp Dermatol. 1988; 13(6): 408-410.
- Kim JW, Park JH, Lee D, Hwang SW, Park SW: Vegetative pyoderma gangrenosum in Behçet’s disease . Acta Derm Venereol. 2007; 87: 365-7.
- Yusuke Kashiwado, Ayumi Uchino, Toshiyuki Ota, Shuji Nagano. Intestinal Behçet’s disease with pyoderma gangrenosum successfully treated with the combination therapy of adalimumab and glucocorticoids. Mod Rheumatol. Early Online. 2016; 1-5.
- Armas JB, Davies J, Davis M, et al Atypical Behcet’s disease with peripheral erosive arthropathy and pyoderma gangrenosum. Clin Exp Rheumatol. 1992; 10(2): 177 180.
- Rustin MH, Gilkes JJ, Robinson TW Pyoderma gangrenosum associated with Behcet’s disease: treatment with thalidomide. J Am Acad Dermatol. 1990; 23(5-1): 941-944.
- Ahronowitz I, Harp J, Shinkai K Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012; 13(3): 191-211.
- Conrad C, Truëb RM. Pyoderma gangrenosum. J Dtsch Dermatol Ges. 2005; 3: 334-42.
- Akay N, Boyvat A, Heper AO, Soykan I, et al. Behcet’s diseaselike presentation of bullous pyoderma gangrenosum associated with Crohn’s disease. Clin Exp Dermatol. 2006; 31: 384-6.
- Ozuguz P, Kacar SD, et al: Genital ulcerative pyoderma gangrenosum in Behçet’s disease: a case report and review of the literature. Indian J Dermatol. 2015; 60: 105.
- M. Cherif, et al. Maladie de Behçet et pyoderma gangrenosum: une association exceptionnelle. La Revue de Médecine Interne. 2023; 44(1): 150-151.
- Ahmadi S, Powell FC. Pyoderma gangrenosum: uncommun presentations. Clin Dermatol. 2005; 23: 612-20.
- Nakamura T, Yagi H, Kurachi K, Suzuki S, Konno H Intestinal Behcet’s disease with pyoderma gangrenosum: a case report. World J Gastroenterol. 2006; 12(6): 979-981.
- Hali F, Khadir K, Chiheb S, Bouayad K, et al Pyoderma gangrenosum and Behcet’s disease: a study of two pediatric cases. Arch Pediatr. 2011; 18(12): 1320-1323.
- Chariatte N, Lysitsa S, Lombardi T, Samson J, editors. Pyoderma gangrenosum (2ème partie): manifestations buccales et présentation d’un cas. 2011.
- Chariatte N, Lysitsa S, Lombardi T, Samson J, editors. Pyoderma gangrenosum (1iere partie): mise au point. Med Buccale Chir Buccale. 2011;17: 121-131.
- Callen JP, Jackson JM. Pyoderma gangrenosum: an update. Rheum Dis Clin North Am. 2007; 33: 787-802.