
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Schnitzler syndrome: A rare disease with neutrophilic dermatosis and monoclonal gammopathy
Saikat Mondal1; Naveen Vairamoorthy1; Jyoti Kotwal1; Jasjit Singh2*
1Department of Hematology, Sir Ganga Ram Hospital, New Delhi, India.
2Department of Clinical Hematology, Sir Ganga Ram Hospital, New Delhi, India.
*Corresponding Author : Jasjit Singh
Department of Clinical Hematology, Sir Ganga Ram
Hospital, New Delhi, India.
Email: jjsingh64@yahoo.co.in
Received : Feb 15, 2024
Accepted : Mar 04, 2024
Published : Mar 11, 2024
Archived : www.jcimcr.org
Copyright : © Singh J (2024).
Abstract
Schnitzler syndrome is a rare chronic auto-inflammatory disease characterized by neutrophilic dermatosis and a monoclonal gammopathy of IgM type. The clinical features of this disease like fever, rash, arthralgias and lymphadenopathy often overlaps with the presenting symptoms of other diseases like Castleman’s disease, adultonset Still’s disease and lymphomas. But the line of treatment is different for each of these clinical conditions. Schnitzler syndrome is associated with over-production of IL-1 and therapies are directed against this interleukin. Here, we present a case of Schnitzler syndrome, who was initially diagnosed as multi-centric Castleman’s disease and was treated with only partial resolution of symptoms.
Keywords: Schnitzler syndrome; Rash; Neutrophilic dermatosis; Gammopathy.
Citation: Mondal S, Vairamoorthy N, Kotwal J, Singh J. Schnitzler syndrome: A rare disease with neutrophilic dermatosis and monoclonal gammopathy. J Clin Images Med Case Rep. 2024; 5(3): 2914.
Introduction
Schnitzler syndrome is a rare acquired chronic auto-inflammatory disease. It is characterized by a pruritic rash, fever, arthralgias and a monoclonal IgM gammopathy, often associated with the overproduction of Interleukin-1 (IL-1). Activating mutations of NLRP3 inflammasome is associated with this condition. The diagnostic criteria of Schnitzler syndrome requires fulfillment of two obligate criteria including a chronic urticarial rash of neutrophilic dermatosis, along with a monoclonal gammopathy of IgM type. The minor criteria includes recurrent fever, arthralgias, lymphadenopathy, organomegaly, leukocytosis and/or elevated ESR along with objective findings of abnormal bone remodeling. At least two of the minor criteria needs to be fulfilled if IgM gammopathy is present, and three minor criteria if IgG gammopathy is present. About 15% to 20% patients will develop a lymphoma, similar to patients of monoclonal IgM gammopathies of undetermined significance [1]. Secondary amyloidosis if present, leads to poorer outcomes in untreated patients [2,3].
Case
A 67 year old female with hypertension, has presented to us with an intermittent lowgrade fever for last 7 to 8 months associated with a significant weight loss of 5-6 kg in the same duration. Fever was accompanied by a generalized, self-resolving reddish maculo-papular rash associated with mild pruritus. On examination, she had pallor along with a palpable left axillary lymph node (largest being of size 2.3x2.5 cm) with no hepatosplenomegaly.
She had earlier undergone a biopsy of this lymph node, and the morphology was suggestive of idiopathic multi-centric Castleman’s disease (plasma cell type). IHC was negative for HHV-8. Her bone marrow aspiration and biopsy showed a 10% increase in plasma cells with normal trilineage hematopoiesis with no increase in blasts nor any granuloma. The plasma cells were CD138 positive and showed mild kappa restriction. Her CT thorax and abdomen showed generalized lymphadenopathy with no organomegaly. Her CBC showed a hemoglobin of 8.9 gm/dl along with neutrophilic leucocytosis of 11,240/uL (polymorphs 73%, lymphoctes 18%) and a normal platelet count of 236,000/uL. Her RBCs were normocytic and normochromic. Her LFT and KFT was normal. Her ESR was 140 mm/hr, serum globulins were raised to 4.8 gm/dl and CRP was 129 mg/dl. Her HIV, anti-HCV and HBsAg were negative. Her serum iron was 63 ug/dl; Tsat 27.89%; ferritin 324 ng/ml; folic acid 12 ng/ml; vitamin B12 <109 pg/ml. Her serum electrophoresis showed a ‘M’ band of 1.39 gm/dl and serum immunofixation showed a monoclonal band corresponding to IgM-kappalane. Her ANA-IF, ANA-profile and RF was negative. Her PET-CT showed multiple FDG avid lymph nodes on both the sides of the diaphragm with no bony lesions. Multi-centric Castleman’s disease (plasma cell type) was diagnosed and she was started on weekly doses of 375 mg/m2 injection rituximab.
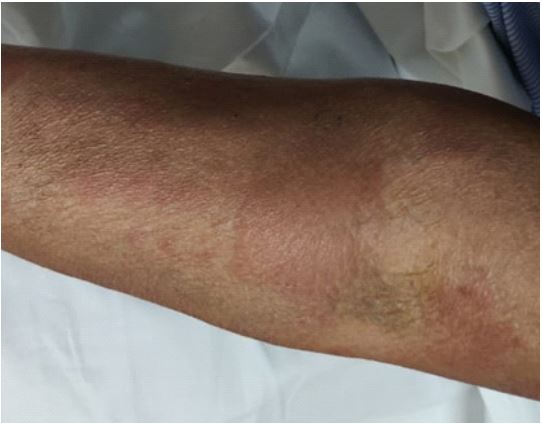
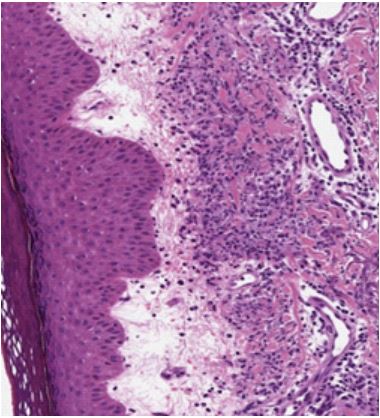
Her fever subsided following two doses of rituximab injection and she was discharged. However, she again presented to us after 3-4 weeks duration with fever and reappearance of the urticarial maculo-papular rash. Her lymph nodes showed a mild decrease in size. Skin biopsy of the rash showed abundant dermal neutrophilic infiltrates, consistent with neutrophilic urticarial dermatosis and a possibility of Schnitzler syndrome was given. There was no evidence of vasculitis or dermal edema. The patient was started on oral steroids along with mycophenolate mofetil, following which her symptoms resolved completely.
Discussion
Schnitzler syndrome is usually associated with severe deterioration in the quality of life. Patient usually presents with fever, arthralgias and a pruritic rash. One-third of these patients develop a lymphoproliferative disorder, mainly Waldenstrom macroglobulinemia. This rate of conversion to lymphomas is almost similar to those of IgM-MGUS cases.
Idiopathic multicentric Castleman’s disease is mostly associated with POEMS symdrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy and Skin lesions). The mandatory criteria of POEMS syndrome was not being fulfilled in our patient. Waldenstrom macroglobulinemia presenting with a IgM monoclonal gammopathy, is often associated with severe hyperviscocity symptoms. IgM myeloma is a rare condition, usually associated with renal involvement and hypercalcemia from osteolytic bony lesions. Our patient, despite of having a IgM-kappa monoclonal gammopathy did not have any symptoms of hyperviscosity like dizziness, headache or blurring of vision nor did she have any peripheral nephropathy. Her PET-CT showed no evidence of any osteolytic lesions. Moreover, this condition needs to be differentiated from other neutrophilic urticarial dermatosis like Adult Onset Still’s Disease (AOSD), which is usually associated with a daily evening flare-up of macula-papular rashes and a very high ferritin levels. AOSD is associated with elevated levels of liver enzymes and often accompanies sore throat, according to Yamagushi criteria. But usually no monoclonal gammopathy is seen in AOSD. Treatment of Schnitzler syndrome includes mostly IL-1 receptor inhibitors like anakinra, canakinumab and rilonacept. Other therapies includes corticosteroids, rituximab, mycophenolate mofetil and cylophosphamide. Our patient was started on immune-suppressive therapies in the form of steroids and MMF, following which her symptoms completely resolved.
Conclusion
Schnitzler syndrome is a rare auto-inflammatory condition, presenting with chronic neutrophilic dermatosis and monoclonal gammopathy. It shows clinical features overlapping with other disease conditions however, treatment of this condition is different from the other related conditions.
Conflict of interest: No conflict of interest to disclose.
References
- Kyle RA, Therneau TM, Rajkumar SV, Remstein ED, Offord JR, Larson DR, Plevak MF, Melton LJ. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003; 102(10): 3759-64. doi: 10.1182/blood-2003-03-0801.
- Claes K, Bammens B, Delforge M, Evenepoel P, Kuypers D, Vanrenterghem Y. Another devastating complication of the Schnitzler syndrome: AA amyloidosis. Br J Dermatol. 2008; 158(1): 182-4.
- de Koning HD, Bodar EJ, van der Meer JW, Simon A. Schnitzler Syndrome Study Group. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007; 37(3): 137-48. doi: 10.1016/j.semarthrit.2007.04.001.