
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Series - Open Access, Volume 5
Two novel ENG variants in families with type 1 hereditary haemorrhagic telangiectasia
Dillon Mintoff1,2; Nikolai P Pace3,4; Nicola Carroll5; Jenna Jenkinson5; Isabella Borg1,3,6*
1Department of Pathology, Faculty of Medicine and Surgery, University of Malta, Malta.
2Department of Dermatology, Mater Dei Hospital, Malta.
3Centre for Molecular Medicine and Biobanking, University of Malta, Malta.
4Department of Anatomy, Faculty of Medicine and Surgery, University of Malta, Malta.
5Molecular Genetics Laboratory, South East Scotland Genetic Service, Western General Hospital, Edinburgh, Scotland.
6Medical Genetics Unit, Pathology Department, Mater Dei Hospital, Malta.
*Corresponding Author : Isabella Borg
Department of Pathology, Faculty of Medicine and Surgery, University of Malta, Malta.
Email: Isabella.borg@um.edu.mt
Received : Mar 05, 2024
Accepted : Mar 28, 2024
Published : Apr 04, 2024
Archived : www.jcimcr.org
Copyright : © Borg I (2024).
Abstract
Hereditary Haemorrhagic Telangiectasia (HHT) is a rare autosomal dominant genodermatosis characterised by cutaneous and visceral telangiectasia, recurrent epistaxis, and Arterio-Venous Malformations (AVMs). In this manuscript, we describe two novel ENG variants in Maltese-Caucasian patients presenting with a variable inter- and intrafamilial clinical phenotypes. This report highlights the importance of diagnostic suspicion of HHT in the presence of cutaneous and/or mucosal telangiectasia and/or AVMs.
Keywords: Hereditary Haemorrhagic Telangiectasia (HHT); Osler-weber-rendu; ENG.
Citation: Mintoff D, Pace NP, Carroll N, Jenkinson J, Borg I. Two novel ENG variants in families with type 1 hereditary haemorrhagic telangiectasia. J Clin Images Med Case Rep. 2024; 5(4): 2963.
Introduction
HHT, also known as Osler-Weber-Rendu syndrome, affects an estimated 1:5,000-1:10,000 persons [1,2]. A diagnosis of HHT is based on clinical criteria brought forward by Shovlin and colleagues (Curaçao criteria) 2 namely, multiple mucocutaneous or visceral telangiectasia, recurrent epistaxis (especially night-time bleeds), visceral Arterio-Venous Malformations (AVMs) and a first degree relative with a diagnosis of HHT. Herein, we report two novel ENG variants in multiple patients of Maltese Caucasian ethnicity suffering from HHT.
Case reports
Proband 1
A 27-year-old female was referred to the genetics clinic in view of a history of epistaxis, mucocutaneous telangiectasia and a pulmonary AVM which was diagnosed during investigation for postpartum hypoxia. Her past medical history was significant for rectal bleeding in the setting of endoscopically detected telangiectasia of the gastric antrum and the duodenum. Having satisfied three of Curaçao’s four diagnostic criteria, the patient was clinically diagnosed with HHT (Table 1).
Proband 2
A 34-year-old male was referred to the genetics clinic after being found to have a dominant arteriovenous fistula in the lower lobe of the right lung, receiving pulmonary arterial supply from the right lower lobe pulmonary artery and draining via the pulmonary vein inserting directly into the left atrium. Smaller fistulae were noted in the apical segment of the upper and lower lobes of the right lung. The patient also had a right-sided aortic arch as well as an aberrant left subclavian artery. These complex radiological findings raised the suspicion for a diagnosis of HHT. Relevant clinical history was significant for epistaxis and examination revealed telangiectasia on the tongue. Having satisfied three of Curaçao’s criteria 2, a clinical diagnosis of HHT was made (Table 1).
Cascade studies
In both probands, targeted capture and sequencing of an HHT gene panel (ACVRL1, ENG, EPHB4, GDF2, RASA1 and SMAD4) was performed using a custom-designed Twist Bioscience panel for library construction and enrichment, and an Illumina MiSeq® platform for paired-end DNA sequencing. Variants of interest were validated using Sanger sequencing. DNA was also analysed using the MLPA P093-C2 and P158-D1 kits, which measure the copy number of the coding exons of the ACVRL1, ENG and SMAD4 genes.
In proband 1, an ENG missense variant c.1280T>G (p.Val427Gly) in exon 10 was identified. A Sanger trace demonstrating the variant is shown in (Figure 1a). Cascade testing of relatives at risk was carried out. The proband’s mother, maternal aunt and cousin were found to harbour the variant. Their clinical, genetic and investigational results are presented in Table 1.
In proband 2, the ENG sequence variant c.1583del (p.Pro528fs) in exon 12 was identified. A Sanger trace demonstrating the variant is shown in (Figure 1b). Cascade testing of relatives at risk was carried out. The proband’s brother, son and daughter were all found to harbour the variant. Their clinical, genetic and investigational results are presented in (Table 1).
Apart from epistaxis in the proband’s father, no other of Curaçao criteria for a diagnosis of HHT were met by the proband’s parents. Nonetheless, bearing in mind that HHT mode of inheritance is Autosomal Dominant (AD), the proband’s father (who experienced epistaxis) was screened for the familial variant; this was not identified on two separate occasions. His asymptomatic mother turned down targeted genetic testing. The late maternal and paternal grandparents did not fulfil any of Curaçao diagnostic criteria.
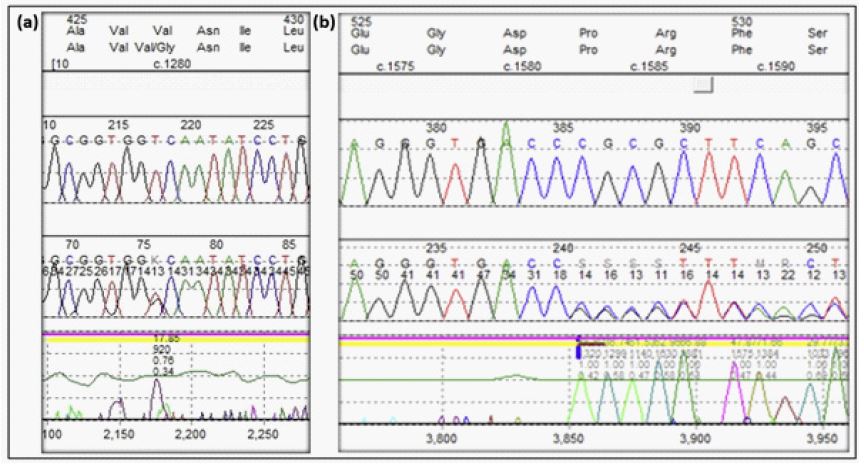
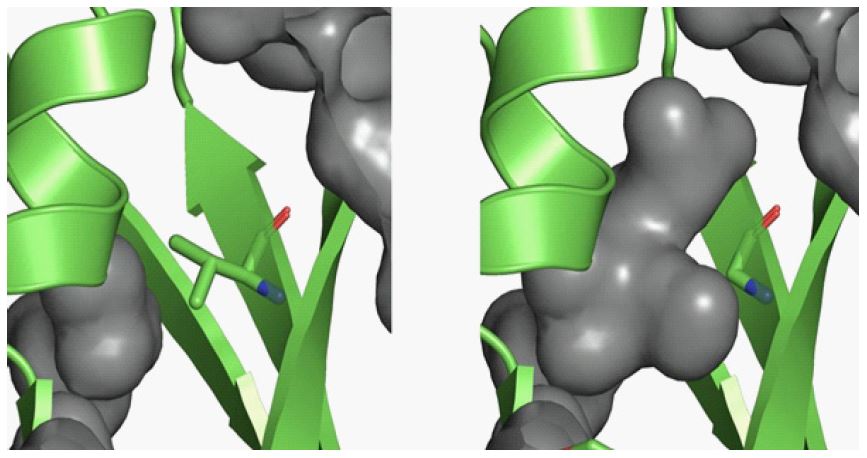
Table 1: Available genetic, clinical and investigational findings of probands and relatives.
| Patient/Relationto proband | VariantIdentified | Ageat Diagnosis | Epistaxis | Telangiectasia | ArteriovenousMalformations | ||||
|---|---|---|---|---|---|---|---|---|---|
| Cutaneous | Mucosal | Visceral | Pulmonary | Cerebral | >Hepatic | ||||
| Family1 Variant:ENG:c.1280T>G (p.Val427Gly) | |||||||||
| Proband(Female) | Yes | 28 | Yes | Yes | Yes | Yes | Yes | Yes | No |
| Mother | Yes | 54 | Yes | No | No | No | Yes | NI | No |
| MaternalAunt | Yes | 42 | Yes | Yes | No | No | No | No | No |
| MaternalCousin1 | Yes | 18 | Yes | Yes | No | No | NI | NI | NI |
| Family2 Variant: ENG:c.1583del (p.Pro528fs) | |||||||||
| Proband (Male) | Yes | 36 | Yes | No | Yes | No | Yes | No | No |
| Mother | NI | NA | No | No | No | NI | NI | NI | NI |
| Father | No | NA | Yes | No | No | No | NI | No | NI |
| Brother | Yes | 35 | Yes | Yes | Yes | No | NI | NI | NI |
| Son | Yes | 6 | Yes | Yes | Yes | NI | NI | NI | NI |
| Daughter | Yes | 9 | Yes | Yes | Yes | NI | NI | NI | NI |
NA=Not applicable NI = Not investigated.
Discussion
HHT is a rare autosomal dominant genodermatosis which exhibits locus heterogeneity, with variants in endoglin (ENG) (9q33-q34.1, HHT1: OMIM #187300), Activin-Like Receptor Kinase-1 (ALK-1) (12q.11-q14, HHT2: OMIM #600376) and Mothers Against Decapentaplegic Homolog 4 (MADH4), encoding for SMAD4 (18q21.2, Juvenile polyposis/HHT syndrome; OMIM:17050) accounting for 80%-96% of patients with HHT [3-5]. Other less common variants such as those in an unidentified gene on chromosome 5q31.3-q32 in HHT3 [6] (OMIM #601101), unidentified gene on chromosome 7p14 in HHT4 [7] (OMIM #610655), and GDF2 gene on chromosome 10q11.22 encoding Bone Morphogenic Protein 9 (BMP9) in HHT5 [8] (OMIM #611506), account for the remainder of documented HHT patients [5].
Patients with HHT exhibit a variable clinical phenotype. In the absence of a family history of HHT, epistaxis may be the presenting sign in up to 90% of patients with the eruptions of mucocutaneous telangiectasia lagging 5 to 20 years after the initial nose bleed [9]. Some patients may be diagnosed with HHT whilst being investigated for chronic anaemia (which is present in up to 50% of patients) [10] or after sustaining potentially fatal complications from previously undocumented visceral AVMs.
In this report, we describe two novel ENG variants. Both variants are absent from the genome aggregation database gnomAD as well as from an ethnically matched reference genome collection.
The ENG: c.1280T>G (p.Val427Gly) substitution is classified as a likely pathogenic variant based on PS4_Sup, PM2, PP1, PP3, PP4 (ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020). Several in-silico predictors support a deleterious effect of this substitution. To gain insight into the possible effect of the p.Val427Gly substitution on the stability, structure and function of the ENG protein, a computational approach was applied. Molecular modelling was conducted based on the crystal structure of ENG (PDB entry 5HZV) using different structural bioinformatics tools. The ENG p.Val427Gly substitution is predicted to be structurally damaging by Missense3D [11], as it results in an expansion of protein cavity volume and a switch between the buried and exposed states of the wild type and variant residues. The substitution has destabilising thermodynamic predictions from Dynamut; Δ Δ G= -2.691kcal/mol (using protein structure as predicted by AlphaFold [accession code AF-P17813-F1]) [12].
On the other hand, the ENG: c.1583del (p.Pro528fs) has not been previously published, but has one ClinVar entry. This frameshift indel creates a premature stop codon and is classified as pathogenic based on PVS1, PM2, PP1 (ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020).
Given that the proband’s symptomatic brother was found to be carrying the familial pathogenic indel variant, and in view of the mode of inheritance being AD, it would be expected for either of the parents to harbour the variant. Since the father (whose history of epistaxis could be explained by other medical reasons including a history of hypertension, anticoagulation for a previous transient ischaemic attack and by occupational exposure to industrial spray and dust) did not carry the variant and the possibility of non-paternity was denied, a likely explanation for the origin of the variant is gonadal mosaicism in either of the proband’s parents. The possibility of asymptomatic carriage [13] of the variant in the proband’s asymptomatic mother is unlikely, given that the affected individuals in this family presented with epistaxis and telangiectasia in childhood.
ENG encodes for endoglin (CD105), a 180 kDa transmembrane glycoprotein that binds ligands of the transforming Growth Factor β (TGF β) [14] and Bone Morphogenic Protein (BMP) [15] families. Exons 1-12 of the ENG encode for the large extracellular domain of the protein whilst exons 13 and 14 encode for the transmembrane and intracellular domains of endoglin, with most variants affecting exons 2 to 11 and 13 [16]. Membrane-bound isoforms (the Long [L], the Short [S]) [17] and a soluble (cleaved) forms [18,19] of the protein have been described. Over 400 ENG variants have been identified, of which the commonest are deletion variants [20]. More than 75% of the described variants are classified as pathogenic [20]. It is likely that ENG variants cause HHT by haploinsufficiency [21], as a result of impaired endoglin folding rather than hindered proteomic interactions [15]. Endoglin is involved in the normal functioning of the vascular endothelium as well as other processes such as inflammation [22]. A diagnosis of HHT based on Curaçao diagnostic criteria is highly predictive of patients harbouring pathogenic ENG or ACVRL1 variants [5]. ENG variants have been associated with an HHT phenotype associated with an increased incidence of pulmonary and cerebral vascular malformations in both paediatric [23] and adult [24] patients. On the other hand, hepatic vascular malformations are less likely in patients with ENG variants when compared with patients harbouring ACVRL1 variants [25]. Patients having ENG variants are likely to experience epistaxis earlier and more severely than patients with other types of HHT-associated variants [25] and are more likely to be diagnosed earlier than patients with ACVRL1 variants [24]. Patients who are actively screened and treated for potential HHT-related visceral complications have been found to have a lifespan comparable to non-HHT patients, justifying this approach [26].
Conclusion
In this report we describe two novel ENG variants thereby expanding the mutational spectrum of this rare genodermatosis. No definitive genotype-phenotype associations have been established, as affected patients exhibit a variable inter- and intrafamilial clinical phenotypes. HHT can be suspected by the clinical identification of any of Curaçao clinical criteria thus making an early diagnosis possible.
Declarations
Funding sources: Nil.
Conflict of interest: Nil to disclose.
Acknowledgement: The patients in this manuscript have given written informed consent to publication of their case details.
References
- McDonald J, Bayrak-Toydemir P, Pyeritz RE. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genetics in Medicine. 2011; 13(7): 607-616. doi:10.1097/GIM.0b013e3182136d32.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000; 91(1): 66-67. doi:10.1002/(sici)1096-8628(20000306)91:1< 66::aid-ajmg12>3.0.co;2-p.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: Current views on genetics and mechanisms of disease. J Med Genet. 2006; 43(2): 97-110. doi:10.1136/jmg.2005.030833.
- McDonald J, Wooderchak-Donahue W, VanSant Webb C, Whitehead K, Stevenson DA, et al. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front Genet. 2015; 6. doi:10.3389/fgene.2015.00001.
- McDonald J, Bayrak-Toydemir P, DeMille D, Wooderchak-Donahue W, Whitehead K. Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia is highly predictive of a pathogenic variant in ENG or ACVRL1 (HHT1 and HHT2). Genetics in Medicine. 2020; 22(7): 1201-1205. doi:10.1038/s41436-020-0775-8.
- Cole SG, Begbie ME, Wallace GMF, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005; 42(7): 577-582. doi:10.1136/jmg.2004.028712.
- Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006; 140(20): 2155-2162. doi:10.1002/ajmg.a.31450.
- Wooderchak-Donahue WL, McDonald J, O’Fallon B, et al. BMP9 Mutations Cause a Vascular-Anomaly Syndrome with Phenotypic Overlap with Hereditary Hemorrhagic Telangiectasia. The American Journal of Human Genetics. 2013; 93(3): 530-537. doi:10.1016/j.ajhg.2013.07.004.
- Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: A clinical analysis. J Med Genet. 1992; 29(8): 527-530.
- Kasthuri RS, Montifar M, Nelson J, et al. Prevalence and predictors of anemia in hereditary hemorrhagic telangiectasia. Am J Hematol. Published online. 2017. doi:10.1002/ajh.24832.
- Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, et al. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J Mol Biol. 2019; 431(11): 2197-2212. doi:10.1016/j.jmb.2019.04.009.
- Rodrigues CH, Pires DE, Ascher DB. DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018; 46(W1): W350-W355. doi:10.1093/nar/gky300.
- Anderson E, Sharma L, Alsafi A, Shovlin CL. Pulmonary arteriovenous malformations may be the only clinical criterion present in genetically confirmed hereditary haemorrhagic telangiectasia. Thorax. 2022; 77(6): 628-630. doi:10.1136/thoraxjnl-2021-218332.
- Ríus C, Smith JD, Almendro N, et al. Cloning of the promoter region of human endoglin, the target gene for hereditary hemorrhagic telangiectasia type 1. Blood. 1998; 92(12): 4677-4690.
- Saito T, Bokhove M, Croci R, et al. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017; 19(9): 1917-1928. doi:10.1016/j.celrep.2017.05.011.
- Olivieri C, Pagella F, Semino L, et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. Journal of Human Genetics. 2007; 52(10): 820-829. doi:10.1007/s10038-007-0187-5.
- Bellón T, Corbí A, Lastres P, et al. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur J Immunol. 1993; 23(9): 2340-2345. doi:10.1002/eji.1830230943.
- Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006; 12(6): 642-649. doi:10.1038/nm1429.
- Hawinkels LJAC, Kuiper P, Wiercinska E, et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010; 70(10): 4141-4150. doi:10.1158/0008-5472.CAN-09-4466.
- ARUP Scientific Resource for Research and Education: ENG Database University of Utah. Accessed. 2020. https://arup.utah.edu/database/ENG/ENG_welcome.php.
- Paquet ME, Pece-Barbara N, Vera S, et al. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum Mol Genet. 2001; 10(13): 1347-1357. doi:10.1093/hmg/10.13.1347.
- Rossi E, Sanz-Rodriguez F, Eleno N, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. 2013; 121(2): 403-415. doi:10.1182/blood-2012-06-435347.
- Kilian A, Latino GA, White AJ, et al. Genotype-Phenotype Correlations in Children with HHT. J Clin Med. 2020; 9(9). doi:10.3390/ jcm9092714.
- Sánchez-Martínez R, Iriarte A, Mora-Luján JM, et al. Current HHT genetic overview in Spain and its phenotypic correlation: data from RiHHTa registry. Orphanet Journal of Rare Diseases. 2020; 15(1): 138. doi:10.1186/s13023-020-01422-8.
- Karlsson T, Cherif H. Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: experience from the Center for Osler’s Disease, Uppsala University Hospital. Upsala Journal of Medical Sciences. 2018; 123(3):153-157. doi:10.1080/03009734.2018.1483452.
- De Gussem EM, Kroon S, Hosman AE, et al. Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. Journal of Clinical Medicine. 2020; 9(11): 3581. doi:10.3390/jcm9113581.