
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
An unusual diagnosis of small bowel bleeding: A case of hereditary hemorrhagic telangiectasia
Li-Han Weng1; Chang-Cheng Su1; Ching-Pin Lin1,2*
1Department of Internal Medicine, Division of Gastroenterology and Hepatology, Chung Shan Medical University Hospital, Taiwan.
2Institute of Microbiology and Immunology, Chung Shan Medical University, Taichung, Taiwan.
*Corresponding Author : Ching-Pin Lin
Department of Internal Medicine, Division of Gastroenterology and Hepatology, Chung Shan Medical University Hospital, Taiwan.
Email: a0982224141@gmail.com
Received : Mar 15, 2024
Accepted : Apr 08, 2024
Published : Apr 15, 2024
Archived : www.jcimcr.org
Copyright : © Lin CP (2024).
Abstract
In clinical practice, small bowel bleeding is primarily attributed to angiodysplasia associated with chronic kidney disease. Hereditary Hemorrhagic Telangiectasia (HHT) represents a relatively rare condition, which poses diagnostic challenges. Herein, we present the case of a 49-year-old male with chronic gastrointestinal bleeding. The diagnosis was established based on the patient’s medical history, imaging findings, and genetic testing.
Keywords: Gastrointestinal bleeding; Hereditary hemorrhagic telangiectasia; Double balloon enteroscopy; Genetic test.
Citation: Weng LH, Su CC, Lin CP. An unusual diagnosis of small bowel bleeding: A case of hereditary hemorrhagic telangiectasia. J Clin Images Med Case Rep. 2024; 5(4): 2987.
Introduction
Hereditary Hemorrhagic Telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal dominant genetic disorder characterized by the development of abnormal blood vessels, leading to Arteriovenous Malformations (AVMs) and telangiectasias. These vascular anomalies can occur in various organs, including the skin, mucous membranes, lungs, brain, and gastrointestinal tract, leading to a range of symptoms such as spontaneous bleeding, anemia, and organ dysfunction.
Case presentation
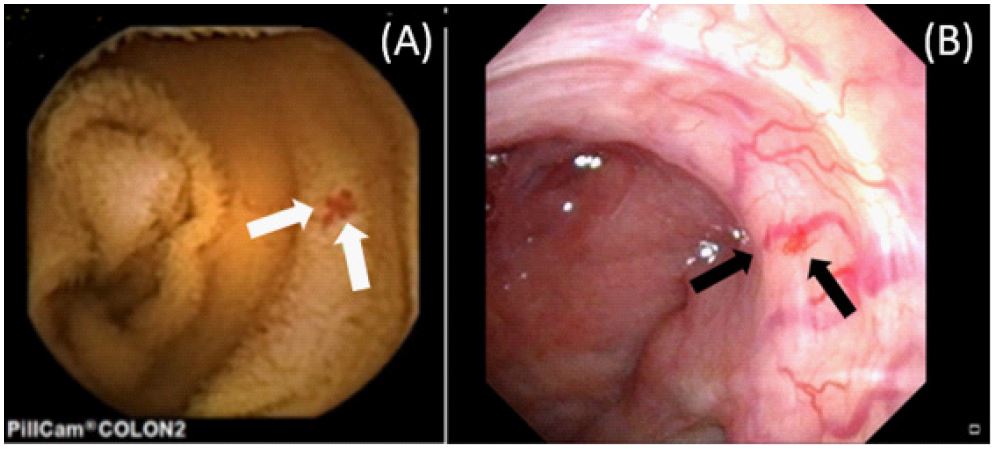
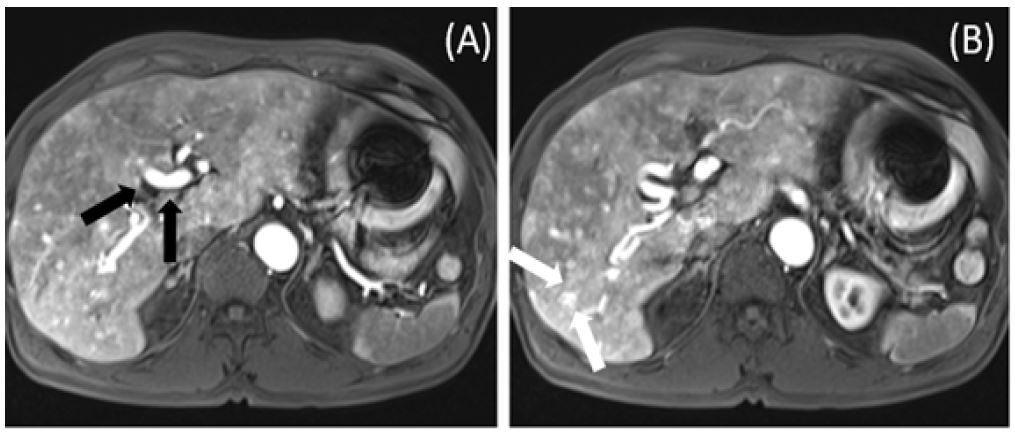
A 49-year-old male presented with intermittent melena over three months. Clinical examination revealed pallor, digit hemorrhagic macules, and a history of epistaxis, gout, and controlled hypertension. The patient reported no medications for bleeding diathesis, had a 30-year history of smoking, and consumed alcohol moderately. There was no significant family history. Biochemistry results indicated microcytic anemia with iron deficiency but no tendency for bleeding. Esophagogastroduodenoscopy and colonoscopy findings were unremarkable. However, Tc-99m RBC scintigraphy demonstrated radioactivity extravasation moving to the central abdomen. Subsequently, capsule endoscopy revealed angiodysplasia with recent bleeding in the proximal jejunum (Figure 1A). Suspecting hereditary hemorrhagic telangiectasia, flexible nasal endoscopy and contrast-enhanced MRI of the brain and abdomen were performed, revealing dilation of nasal capillaries and angiodysplasia (Figure 1B) as well as hepatic arteriovenous malformations (Figure 2). Genetic testing identified a missense variant in the ACVRL1 gene. The patient was started on thalidomide therapy (100 mg daily), with continuous monitoring.
Discussion
Hereditary Hemorrhagic Telangiectasia (HHT), also known as Osler-Web-Rendu syndrome, is an autosomal dominant disorder characterized by abnormal blood vessel formation. This leads to large visceral Arteriovenous Malformations (AVMs) in the brain, lungs, and liver, and smaller telangiectasias at characteristic mucocutaneous sites (lips, nose, and fingers) as well as in the gastrointestinal tract. The prevalence of HHT is similar in Japan, Europe, and the U.S., approximately 1:8,000. The clinical diagnostic criteria, known as the Curaçao criteria, established in 2000, are strongly predictive of genetic confirmation [1]. Most pathogenic mutations associated with HHT are null alleles in the ENG, ACVRL1, and SMAD4 genes, which encode proteins expressed in endothelial cells involved in the BMP/TGF-β pathways [2]. Emerging evidence has also highlighted the involvement of BMP9 (GDF2) and BMP10 [3].
The Second International Guidelines for Hereditary Hemorrhagic Telangiectasia (HHT) [4] underscore a comprehensive approach for managing various manifestations associated with this condition. For adults with confirmed or potential HHT, MRI screenings are recommended for detecting brain Vascular Malformations (VMs), utilizing both contrast and non-contrast protocols to maximize sensitivity. In cases of acute brain VM hemorrhage, immediate treatment at a neurovascular specialist center is essential. Additionally, all adults with brain VMs, barring emergency bleeding situations, should seek evaluation and personalized management at neurovascular expertise centers. Pregnant women with HHT and asymptomatic brain VMs are advised to delay VM treatment until postpartum, following standard obstetrical practices for delivery.
When it comes to liver VMs in HHT patients, especially those exhibiting symptoms of complications like heart failure or pulmonary hypertension, diagnostic assessments including Doppler ultrasound, contrast-enhanced CT, or MRI are crucial. Intensive management is reserved for those with symptomatic and complicated liver VMs, recommending co-management with specialized HHT centers for cases involving high-output cardiac failure and pulmonary hypertension. For patients with symptomatic high-output cardiac failure from liver VMs not responding to initial treatments, intravenous bevacizumab is considered. Liver transplantation may be indicated for severe complications. However, liver biopsy and hepatic artery embolization are discouraged due to their risks and temporary benefits.
For pulmonary AVMs, the initial diagnosis involves transthoracic contrast echocardiography. Treatment primarily involves transcatheter embolotherapy. Patients are advised to follow long-term precautions including antibiotic prophylaxis for bacteremia risk procedures, careful intravenous line management to avoid air embolism, and avoiding SCUBA diving. Regular follow-up is imperative to monitor for the progression of untreated AVMs and the potential reperfusion of treated AVMs.
Addressing HHT-related epistaxis begins with moisturizing topical therapies for the nasal mucosa. If ineffective, oral tranexamic acid is the next recommendation. For non-responsive cases, options include ablative therapies such as laser treatment and electrosurgery, or more advanced treatments like systemic antiangiogenic agents. Dermaplasty and nasal closure are considered for unresponsive severe epistaxis. Preventative measures and referral to otorhinolaryngologists with HHT expertise are advised for comprehensive care.
Lastly, for suspected HHT-related bleeding, esophagogastroduodenoscopy serves as the primary diagnostic tool, with colonoscopy recommended for those at risk for colorectal cancer or suspected of having SMAD4-HHT. Capsule endoscopy may be utilized when initial screenings are inconclusive. The severity of HHT-related gastrointestinal bleeding is categorized based on the patient’s response to iron supplementation, guiding the treatment strategy from oral antifibrinolytics to intravenous bevacizumab for severe cases. These guidelines emphasize a methodical and specialized approach to managing HHT, aiming to improve outcomes and quality of life for affected individuals.
Conclusion
The diagnosis of Hereditary Hemorrhagic Telangiectasia (HHT) should be considered in cases of recurrent small intestinal or nasal bleeding. Unfortunately, this condition is often overlooked in clinical practice due to its relative rarity. We report this case with the aim of increasing awareness and vigilance among clinicians regarding HHT.
Declarations
Data availability: Data can be provided on reader’s request.
Contributors: Li-Han Weng; Chang-Cheng Su; Ching-Pin Lin
Patient consent: Obtained.
Financial support and sponsorship: Nil.
Conflicts of interest: There are no conflicts of interest.
Acknowledgements: We would like to thank for De-Fu Weng, MD, providing the help on genetic test.
References
- Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, Nozaki J, Inoue S, & Koizumi A. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002; 19(2): 140-148. https://doi.org/10.1002/humu.10026.bvcx.
- David L, Mallet C, Mazerbourg S, Feige JJ, & Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2006; 109(5); 1953-1961. https://doi.org/10.1182/blood-2006-07-034124.
- McDonald J, Bayrak-Toydemir P, DeMille D, Wooderchak-Donahue W, & Whitehead K. Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia is highly predictive of a pathogenic variant in ENG or ACVRL1 (HHT1 and HHT2). Genetics in Medicine. 2020; 22(7): 1201-1205. https://doi.org/10.1038/s41436-020-0775-8.
- Faughnan ME, Mager JJ, Hetts SW, Palda VA, Lang-Robertson K, Buscarini E, Deslandres E, Kasthuri RS, Lausman A, Poetker D, Ratjen F, Chesnutt MS, Clancy M, Whitehead KJ, Al-Samkari H, Chakinala M, Conrad M, Cortes D, Crocione C, Zarrabeitia R. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann Intern Med. 2020; 173(12): 989-1001. https://doi.org/10.7326/m20-1443.