
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Short Report - Open Access, Volume 5
Juvenile overlap syndrome: Considering scleromyositis
Ghita Sqalli Houssini¹*; Hanane Baybay¹; Zakia Douhi¹; Meryem Soughi¹; Sara Elloudi¹; Fatima Zohra Souilmi²; Mustapha Hida²; Fatima Zahra Mernissi¹
1Dermatology Department, Hassan II University Hospital Center, Fez, Morocco.
2Pediatric Department, Hassan II University Hospital Center, Fez, Morocco.
*Corresponding Author : Ghita Sqalli Houssini
Dermatology Department, Hassan II University Hospital Center, Fez, Morocco.
Tel: +21 2655220441.
Email: gsqallihoussini@gmail.com.
Received : Jan 09, 2024
Accepted : Apr 09, 2024
Published : Apr 16, 2024
Archived : www.jcimcr.org
Copyright : © Houssini GS (2024).
Abstract
Scleromyositis refers to a combination of characteristics found in both systemic scleroderma and polymyositis, with identification often based on the presence of the PM-SCL antibody. Currently, there are no well-defined criteria for classifying this syndrome. In childhood cases exhibiting sclerodermoid features, they are often categorized as scleroderma due to the distinct nature of the condition in this age group. We report two cases of children presenting with cutaneous signs of systemic scleroderma and associated myositis. The first case exhibits sclerodactyly, calcinosis, and myalgias with a positive anti-PM/SCL antibody, while the second case shows swollen fingers, clear Raynaud’s phenomenon, and myalgias but a negative anti-PM/SCL antibody. The clinical and biological variation in these cases prompts the dermatologist to consider scleromyositis as a juvenile overlap syndrome, given the sclerodermiform pattern and myositis, regardless of the positivity or negativity of the anti-PM/SCL antibody. As for the treatment, managing Raynaud’s and initiating a low dose of corticosteroid therapy, possibly combined with immunoglobulins, appears to be effective.
Keywords: Scleromyositis; Juvenile; Overlap; Children; PM/SCL; Calcinosis.
Citation: Houssini GS, Baybay H, Douhi Z, Soughi M, Elloudi S, et al. Juvenile overlap syndrome: Considering scleromyositis. J Clin Images Med Case Rep. 2024; 5(4): 2988.
Case report
In the presence of a juvenile overlap syndrome, combining atypical cutaneous signs of systemic sclerosis with muscle involvement, scleromyositis should be considered, despite the absence of anti-PM/SCL antibodies and the lack of cutaneous signs favoring dermatomyositis. We present two cases of children with cutaneous manifestations of systemic sclerosis associated with clinical muscle involvement, with positive anti-PM-SCL antibodies in one case and negative in the other.
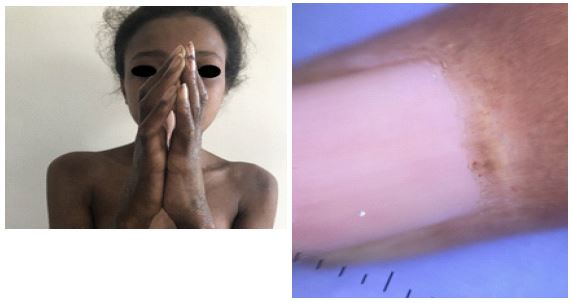
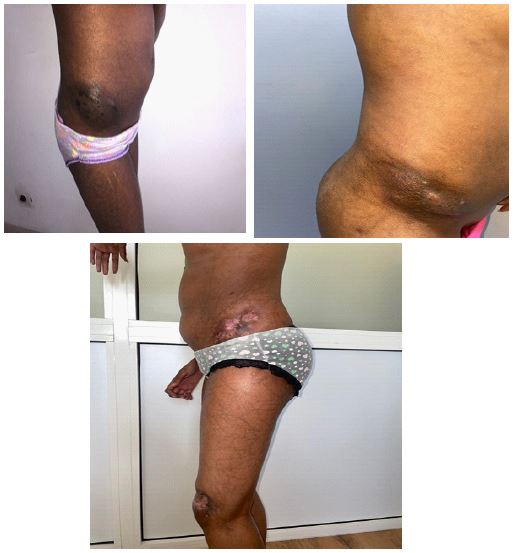
First case: A 13-year-old child exhibited diffuse skin sclerosis and Raynaud’s phenomenon over the past year. Examination revealed sclerodactyly with a positive prayer sign (Figure 1a), a rigid facies, and calcinosis. Periungual dermoscopy showed desolate areas and tortuous capillaries in a fern-leaf pattern indicative of minor dystrophy (Figure 1b). Investigations revealed elevated muscle enzymes, positive anti-PM-SCL antibodies, confirmed myogenic syndrome on Electromyography (EMG), and myositis on muscle biopsy with systemic pulmonary and digestive involvement. The patient received Nicardipine 25 mg/day, Prednisone 7 mg/day, and intravenous immunoglobulins. There was an improvement in sclerosis, and the calcinosis size decreased (Figure 2a-2c).
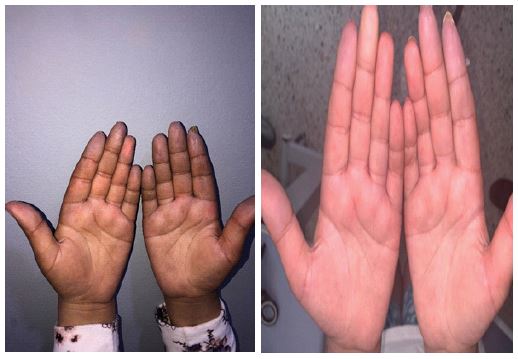
Second case: An 11-year-old child presented with dyschromia affecting almost the entire skin, facial and finger edema, myalgia, and Raynaud’s phenomenon over the past year (Figure 3a). Examination revealed pulpitis with a positive stool sign. Periungual dermoscopy showed desolate areas, and investigations indicated a slight elevation of muscle enzymes with negative anti-PM-SCL antibodies. Thoracic Computed Tomography (CT) revealed esophageal dilation. The child was treated with Nicardipine 0.5 mg/kg/day and Prednisone 7 mg/day. The evolution showed the disappearance of finger edema and improvement in Raynaud’s phenomenon (Figure 3b).
Scleromyositis is a rare overlap syndrome in children, often initially considered systemic sclerosis due to a predominant sclerodermiform pattern, as observed in our cases. Some authors noted skin sclerosis with swollen fingers without sclerodactyly or pulpitis, sparing the face, and with calcinosis less extensive than in case 1. Raynaud’s phenomenon appears late, and periungual dermoscopy does not reveal desolate areas. However, in our cases, pigmentation disorders, sclerodactyly, pulpitis, and desolate areas in periungual dermoscopy were observed. Both cases involved purely muscular forms without cutaneous signs of dermatomyositis. Anti-PM/SCL antibodies allow diagnosis and long-term monitoring but may be lacking, as in case 2.
This suggests individual variation in the clinical characteristics of scleromyositis. A scleroderma-like cutaneous pattern, mainly affecting the hands (swollen or sclerotic fingers) and secondarily the face, appears more frequent than dermatomyositis signs such as Gottron’s papules or periorbital edema. It’s important to note that arthralgia is common, and muscular involvement varies considerably, sometimes clinically imperceptible. Muscle enzymes can be within normal limits, and electromyography may show no abnormalities [4]. Treatment involves low-dose corticosteroids (0.5-1 mg/kg/day), possibly combined with intravenous immunoglobulins for better efficacy, as adopted in our cases [5].
Juvenile scleromyositis is a rare overlap syndrome in children, and there are no predefined criteria for diagnosis. However, the presence of an atypical sclerodermiform pattern associated with dermatomyositis or myositis should raise suspicion. The anti-PM/SCL antibody can guide the diagnosis, but it can be negative in certain cases. Early management is crucial for determining the prognosis in children.
Consent: The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
References
- Błaszczyk M, Jabłońska S, Szymańska-Jagiełło W, Jarzabek-Chorzelska M, Chorzelski T, Mohamed AH. Childhood scleromyositis: an overlap syndrome associated with PM-Scl antibody. Pediatr Dermatol. 1991; 8(1): 1-8.
- Marcus M, Ilyas M, Tolaymat A. Childhood scleromyositis with a negative PM/Scl antibody. Joint Bone Spine. 2010; 77(1): 73-5.
- Loo RJ, Nocton JJ, Harmelink MM, Chiu YE. Anti-Ku antibody-positive systemic sclerosis-polymyositis overlap syndrome in an adolescent. Pediatr Dermatol. 2020; 37(5): 960-961.
- Jablonska S, Chorzelski TP, Blaszczyk M, Jarzabek-Chorzelska M, Kumar V, Beutner EH. Scleroderma/polymyositis overlap syndromes and their immunologic markers. Clin Dermatol. 1992; 10(4): 457-72.
- Abinun M, Estlin E, Gardner-medwin D, Spickett G.P, Mcneela BJ, Cant A.J. Failure of First-Line Therapy with Intravenous Immunoglobulin in a Child with Scleromyosltis. British Journal of Rheumatology. 1996; 35: 1029-1036.