
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Histopathology’s role in the diagnosis of middle ear adenoma
Abdelrahman Osman1; Dalia Y Ibrahim2*
1University of Cincinnati College of Medicine, Cincinnati, OH, USA.
2Department of Pathology, University of Toledo, Toledo, OH, USA
*Corresponding Author : Dalia Y Ibrahim
Department of Pathology, University of Toledo, Toledo, OH, USA.
Tel: 419-383-4511
Email: dalia.ibrahim@utoledo.edu
Received : May 02, 2024
Accepted : May 20, 2024
Published : May 27, 2024
Archived : www.jcimcr.org
Copyright : © Ibrahim DY (2024).
Abstract
Middle ear adenoma is a rare disease that originates, and may extend beyond, the middle ear. It affects patients of a wide age range, has no gender predominance, and has no specific presenting symptoms. Patients typically present with hearing loss, ear fullness, tinnitus, or pain. We report a case of a middle ear adenoma in a 46-year-old woman who complained of progressive right ear problems. She had no symptoms in her left ear. Physical examination was unremarkable in both ears. Imaging could not point to a definite diagnosis. Surgical excision of the mass with tympanoplasty, mastoidectomy, and ossicular chain reconstruction were performed. Histological and immunohistochemical examinations were conducted to confirm diagnosis of middle ear adenoma.
Keywords: Histology; Pathology; Immunohistochemistry; Middle ear; Adenoma.
Abbreviations: MEA: Middle Ear Adenoma; CT: Computed Tomography; CK7: Cytokeratin 7.
Citation: Osman A, Ibrahim DY. Histopathology’s role in the diagnosis of middle ear adenoma. J Clin Images Med Case Rep. 2024; 5(5): 3076.
Introduction
Middle Ear Adenoma (MEA) is a rare disease thought to arise from the middle ear mucosa. Its rarity and unusualness have led to its controversial classification of middle ear masses and a lack of research regarding their cell line of origin, history, and progression. It presents with no specific symptoms, although many patients complain of facial paralysis, unilateral hearing loss, and pain. Patients have ranged from 14 to 80 years of age, with no sex predominance [1]. Imaging usually demonstrates a nonspecific soft tissue mass with no evidence of bone erosion. The lesion may be embedded in the ossicles.
MEA was originally thought to be separate from neuroendocrine tumors, but studies have now shown that MEA can have mixed patterns of differentiation, with exocrine (glandular) and neuroendocrine differentiation representing opposite parts of the spectrum. Clinical diagnosis is very difficult as it has no specific findings clinically nor on imaging. Hence, definitive diagnosis and differentiation of an MEA is dependent on histopathological and immunohistochemical examination. They typically appear white, yellow, gray, reddish-brown, and firm. They are poorly vascularized and may entrap the ossicles [2]. Once found, surgical excision is typically performed with or without reconstruction of hearing [3]. We report the case of an MEA in a 46-year-old female who presented with right ear problems for more than a year.
Case presentation
A 46-year-old white female presented with progressive right ear problems since mid-2019. She complained of a plugged feeling, muffling, tinnitus, and crackling. She denies any ear pain, headaches, or vertigo, and denies any symptoms on the left side.
Upon physical examination, the patient’s right and left ear both presented with no drainage. Neither tympanic membrane was erythematous. Discovery of right-sided conductive hearing loss led to a Computed Tomography (CT) scan, which showed significant right mastoid cell changes as well as a soft tissue inflammatory mass within the middle ear. The mass was consistent with either a cholesteatoma, adenoma, or neurofibroma.
The patient underwent right tympanoplasty with mastoidectomy and ossicular chain reconstruction. During surgery, the mass was noted to fill the entire middle ear. It was soft and ballotable. It seemed to separate from the chorda tympani nerve. The incudostapedial joint was eroded, as the stapes was involved by the tumor pushing it out of the vestibule. There was no involvement of the facial nerve.
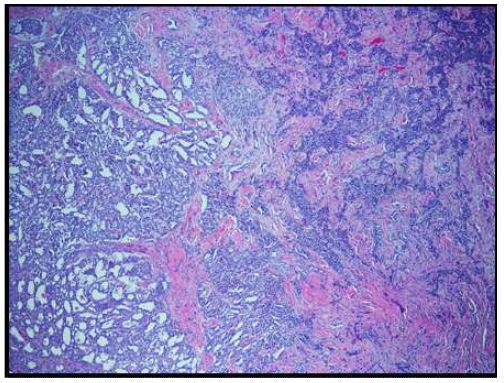
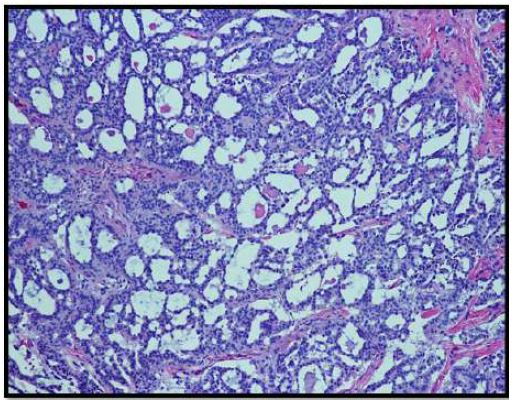
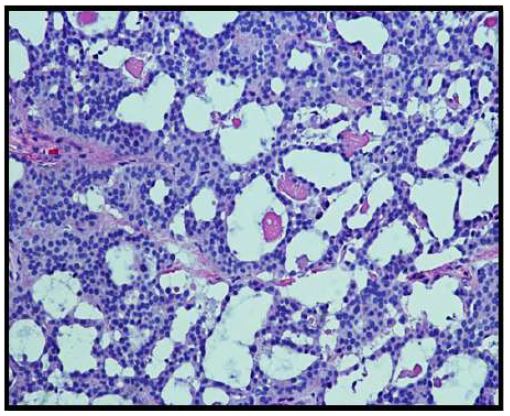
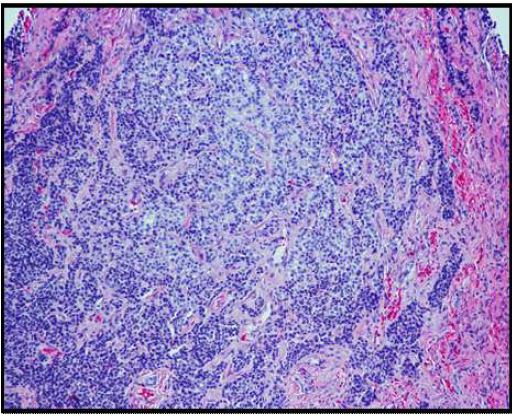
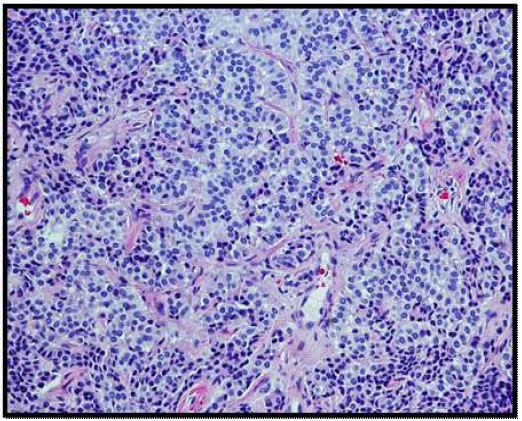
On histopathological examination, the neoplasm (Figure 1) showed mixed pattern with glandular (Figures 2 and 3), trabecular (Figures 4 and 5), and focally solid architecture. There was no necrosis, mitoses, or marked nuclear pleomorphism identified. No perineural or lymphovascular invasion was seen. Tumor ducts showed a dual cell population: Inner, flattened eosinophilic cells and basal cuboidal cells.
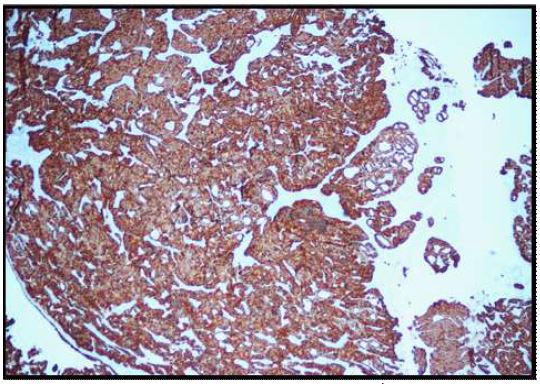
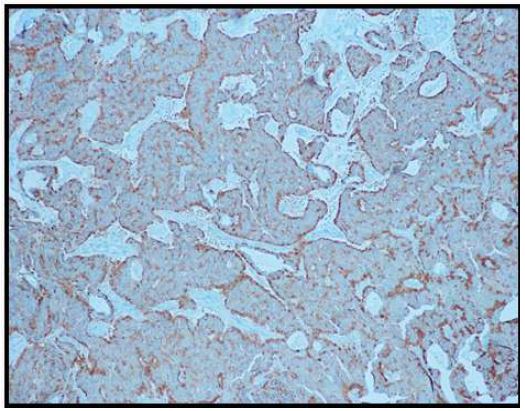
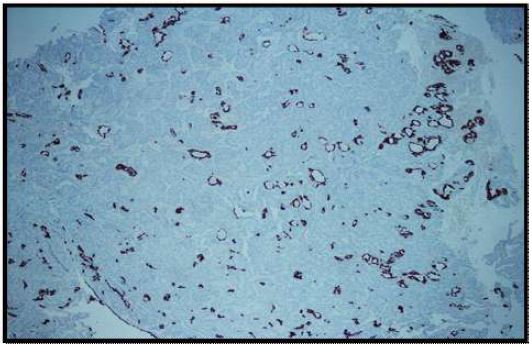
Immunohistochemical stains were performed. Neoplastic cells were positive for AE1/AE3 (Figure 6), synaptophysin (Figure 7), CK 7 (luminal cells in the glandular spaces) (Figure 8), and p63 (basal layer) (Figure 9). The cells were negative for chromogranin, thyroglobulin, and Pax8. The diagnosis was consistent with middle ear adenoma.
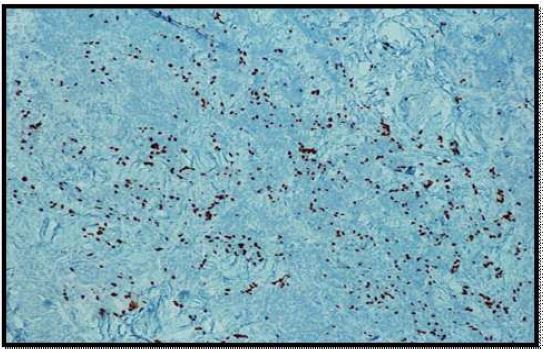
Discussion
MEA is a rare and benign tumor of the middle ear, accounting for 2-4% of all ear tumors [3,4]. Histopathologically, it shows architectural, cytological, and immunohistochemical properties of both neuroendocrine and glandular differentiation. MEA occurs over a wide age range (average of 45 years), has no sex predominance, and no characteristic symptoms. Most patients present with ear fullness, hearing loss, and tinnitus [5]. While a majority of MEA do not invade the facial nerve, some patients also present with facial paralysis. The lesions typically present as white, yellow, gray, or reddish-brown soft masses. They are poorly vascularized and may invade any part of the middle ear, including the Eustachian tube, ossicles, mastoid air spaces, and chorda tympani nerve [6].
On high resolution CT, MEA may present as a soft tissue mass in the middle ear without bone destruction [5]. Differential diagnosis includes congenital cholesteatoma, carcinoid tumor, schwannoma, teratoma, meningioma, and paraganglioma of the middle ear. Fortunately, these masses present differently histologically, and thus definitive diagnosis cannot be completed until surgical resection of the mass is conducted and pathological examination is performed [6]. Upon excision, histologic and immunohistochemical examinations can confirm the presence of an MEA.
The histological patterns of MEA may be solid, glandular, or trabecular. The tumor cells may be cuboidal or columnar. Nuclei are round to oval and assume a “salt and pepper” chromatin appearance. Moderate nuclear pleomorphism may be noted [1]. Papillary features are often not present on histological exams.
Confirmational diagnosis is often completed using immunohistochemical stains. MEA was positive 90% of the time for cytokeratin cocktails, 81% for CAM 5.2, and 90% for CK7. Focally and weakly positive staining may be accomplished with CK20, which was positive 6% of the time. Additionally, differentiation of an MEA may also affect immunohistochemical exams. Neuroendocrine markers such as chromogranin (88% of cases), neuron-specific enolase (50% of cases), synaptophysin (31% of cases), and serotonin (25% of cases) may be found, but not all markers may be present. These typically stain the basal layer of glandular cells [1].
MEA is a difficult condition to diagnose due to its rarity and similarity with other middle ear masses. After surgical resection, patients must be followed up for possible recurrence, although recurrence is rare. Metastasis is also considered extremely rare. Regular follow-up with otoscopy, audiometry, and CT are recommended [5].
Conclusion
Middle ear adenoma is a rare condition. Its scarcity in the medical field has led to controversial classifications and questions regarding its origin, natural history, and progression. Exploration and surgical extraction of these tumors is often performed, and histologic and immunohistochemical examinations are needed for a definitive diagnosis. These tumors can show exocrine and/or neuroendocrine properties. Long-term follow up and observation is recommended, although recurrence and metastasis are rare. Despite being benign, these lesions can be locally destructive. Our case highlights the importance of prompt diagnosis and treatment of these lesions to prevent progression of dysfunction.
References
- Berns S, Pearl G. Middle ear adenoma. Arch Pathol Lab Med. 2006; 130: 1067-1069.
- Zan E, Limb CJ, Koehler JF, et al. Middle ear adenoma: A challenging diagnosis. AJNR Am J Neuroradiol. 2009; 30: 1602-03.
- Isenring D, Pezier TF, Vrugt B, et al. Middle ear adenoma: case report and discussion. Case Rep in Otolaryngol. 2014; 2014: 342125.
- Shujin He, Susu Shi, Rui Qin, et al. Neuroendocrine neoplasms of the middle ear: Report of 2 cases and review of the literature. Int J Clin Exp Pathol. 2019; 12(9): 3453-3458.
- Baku M, Ueda H. A rare case of middle ear adenoma. Nagoya J Med Sci. 2014; 76: 355-360.
- Alzubaidi Y, Abdulsattar J, Al-Delfi F, et al. Middle ear adenoma: A rare case. Am J Clin Pathol. 2018; 149: 142.