
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Myocardial catastrophe in catastrophic antiphospholipid antibody syndrome
Muhammad Khan1*; Adithya Kumar1; Kevin Li1; Jiang Wang2; Umama Gorsi1; Prajakta Phatak1; Wilbert S Aronow3; Manisha Das1
1Department of Internal Medicine, Division of Cardiology, University of Cincinnati, Cincinnati, Ohio, USA.
2Department of Pathology & Lab Medicine, Division of Anatomic Pathology, University of Cincinnati, Cincinnati, Ohio, USA.
3Department of Cardiology, Westchester Medical Center and New York Medical College, USA.
*Corresponding Author : Muhammad Khan
Department of Internal Medicine, Division of Cardiology, University of Cincinnati, Cincinnati, Ohio, USA.
Tel: +1-347-399-4750;
Email: khan2mm@ucmail.uc.edu
Received : May 06, 2024
Accepted : May 22, 2024
Published : May 29, 2024
Archived : www.jcimcr.org
Copyright : © Khan M (2024).
Abstract
Background: Catastrophic Antiphospholipid Syndrome (CAPS) is a rare, life-threatening condition characterized by rapid, widespread thrombosis. Cardiac involvement, most commonly valvulopathy, is documented in up to 53% of cases, but fulminant myocarditis is much rarer. This report highlights a complex case of CAPS with severe cardiac involvement in a young man with a history of SLE and antiphospholipid antibody syndrome, illustrating the diagnostic and therapeutic challenges of this condition.
Case report: A 32-year-old man with SLE and antiphospholipid antibody syndrome presented with facial swelling and abdominal pain, quickly deteriorating in to cardiogenic shock and progressing to multiorgan failure. His acute presentation along with his history of autoimmune conditions raised concern for CAPS which was confirmed with imaging and histopathology.
Conclusion: This case highlights the aggressive nature of CAPS and the importance of prompt, multidisciplinary approach to management. Despite early diagnosis and optimal guidelines directed management, outcomes remain poor. Novel treatments, such as monoclonal antibodies and ventricular assist devices, offer potential avenues for refractory cases.
Citation: Khan M, Kumar A, Li K, Wang J, Gorsi U, et al. Myocardial catastrophe in catastrophic antiphospholipid antibody syndrome. J Clin Images Med Case Rep. 2024; 5(5): 3081.
Introduction
APS is a rare, immune-mediated, acquired hypercoagulable disorder characterized by persistent antiphospholipid antibodies (aPL) in combination with venous, arterial, and microvascular thromboembolism. CAPS is a rare and life-threatening form of APS which is characterized by a) clinical evidence of multiple organ involvement (commonly, three or more organs) developing over a very short period or within a week; b) histopathological evidence of multiple small vessel occlusions, and c) laboratory confirmation of the presence of aPL (including anticardiolipin antibodis, anti-β2 microglobulin, and/or lupus anticoagulant), usually in high titers.
Case presentation
A 32-year-old man with a past medical history of Systemic Lupus Erythematosus (SLE) and antiphospholipid antibody syndrome (APS) presented to the emergency department with a 1-week history of worsening facial swelling and abdominal pain. Physical exam was significant for moderate tachycardia, hypertension, rebound abdominal tenderness, bilateral flank and suprapubic pain, superficial facial swelling, and scleral icterus. While awaiting evaluation, the patient became hemodynamically unstable, requiring inotropic support and mechanical ventilation. Electrocardiogram demonstrated sinus rhythm with right axis deviation, without acute ST segment or T wave changes. Bedside echocardiography revealed a newly reduced ejection fraction of 5-10%. High sensitivity troponin was 823 ng/L, and brain natriuretic peptide was 1074 pg/mL. After stabilization, the patient was admitted to the cardiovascular intensive care unit to investigate potential autoimmune and infectious causes of cardiogenic shock (SCAI Stage C).
The patient was diagnosed with APS and SLE 6 months prior to admission after presenting with polyarthritis and flank pain following a motor vehicle collision. At the time, he was found to have positive autoimmune titers, including anti-cardiolipin IgM, lupus anticoagulant, double-stranded DNA, anti-nuclear antibody, p- and c-ANCA, and rheumatoid factor. β2 microglobulin, MPO, and PR-3 antibodies were negative. CT abdomen and pelvis initially demonstrated renal and splenic lacerations secondary to trauma, but repeat imaging showed multiple foci of new infarction in both organs. Anticoagulation was initiated with apixaban, as the patient was unamenable to starting warfarin, but they were eventually transitioned after repeat imaging showed stable infarcts and was deemed apixaban failure. Hydroxychloroquine and prednisone were also initiated for SLE management. A transesophageal echocardiograph was obtained and demonstrated normal structure and systolic function. The patient had no significant prior medical history or known comorbidities, including a negative family history of autoimmune and cardiac disease.
The initial differential diagnosis included SLE exacerbation/myocarditis, thrombotic microangiopathy, disseminated intravascular coagulation, small vessel vasculitis, including ANCA-associated vasculitis, and septic shock secondary or infection.
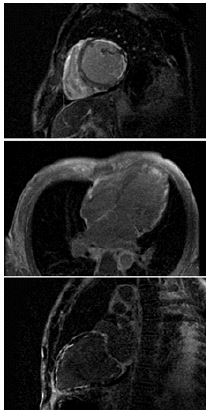
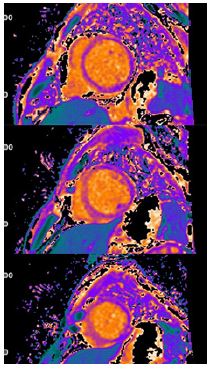
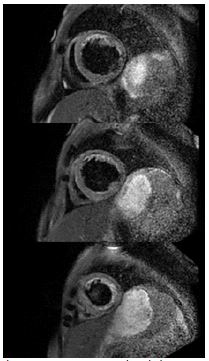
Laboratory studies on admission were significant for an elevated d-dimer, sub-therapeutic INR, lactic acidosis, elevated transaminases, and hyperbilirubinemia. CT pulmonary angiography demonstrated mild pulmonary edema without evidence of pulmonary embolism. CT abdomen pelvis demonstrated stable splenic and renal infarcts, gastric wall thickening, and enhancement of the ileum, concerning for gastritis and enteritis. Complete 2D transthoracic echocardiography revealed severe diffuse hypokinesis with an estimated EF of 10-15%, regional dyskinesia of the apical segment, and a mildly dilated left ventricle cavity (Videos 1 and 2). Cardiac MRI confirmed a dilated left ventricle, global severely reduced EF (Video 3), and further revealed extensive patchy delayed gadolinium enhancement (Figure 1) and abnormal T1 and T2 mapping (Figures 2 and 3). Resting first pass perfusion was normal. Bacterial and viral tests as a cause of myocarditis were unremarkable. The patient was started on pulse dose steroids and cyclophosphamide immediately. Left and right heart catheterizations were performed with biopsy, confirming moderate to severe stenoses of the left anterior descending, left circumflex (Video 4), and right coronary arteries (Video 5). Endomyocardial biopsy was significant for lymphocytic myocarditis without myocyte necrosis.
After stabilization, the patient was extubated, and inotropes and vasopressors were weaned. His clinical course then became immediately complicated by worsening severe abdominal pain, prompting repeat abdominal CT imaging, which revealed multifocal small bowel and colonic infarcts, non-opacification of small bowel and branches of mesenteric arteries, and thrombotic occlusion of the splenic artery. Despite therapeutic anticoagulation, the development of these severe thrombotic complications was consistent with Catastrophic Antiphospholipid Syndrome (CAPS). The rheumatology and hematology services were consulted, and he was initiated on intravenous IgG immunoglobulin (IVIG) and plasmapheresis (PLEX). He was then transferred to the acute care surgical service for emergent exploratory laparotomy and underwent partial gastrectomy, left hemicolectomy, splenectomy, partial pancreatectomy, cholecystectomy, and small bowel resection. Despite all clinical efforts, his condition continued to worsen, requiring reintubation and repeat exploratory laparotomy, which demonstrated additional diffuse ischemia and necrosis of the stomach and small bowel.
Discussion
Approximately 500 cases of CAPS have been recorded in an international registry established by the European Forum of Antiphospholipid Antibodies in 2000. Approximately 70% of patients are female, with a mean age of 39. Nearly 30% had associated SLE, and the overall mortality rate was 36% [1]. Similar to our patient, intraabdominal involvement is generally the first clinical manifestation of CAPS, primarily renal, hepatic, and gastrointestinal disease. Cardiac involvement during catastrophic episodes is reported to be as high as 53%, with the most common manifestations being valvopathy and coronary artery disease [1,2]. Our literature review revealed only a few documented cases of myocarditis secondary to CAPS [3].
The 16th International Congress of Antiphospholipid Antibodies Task Force conditionally recommended triple therapy with anticoagulation, glucocorticoids, and plasmapheresis or IVIG as first-line treatment for CAPS, with evidence supporting improved survival [2]. However, overall survival remains dismal, as low as 36% [6]. 68% of cases are suspected to be precipitated by a trigger such as infection, surgery, or trauma. If a specific stimulus is identified, it should be targeted aggressively and early to prevent further exacerbation.
More recently, several cases have documented the use of monoclonal antibodies such as rituximab and eculizumab as second-line treatment for refractory CAPS, with mixed success [4,5]. Although these drugs have demonstrated efficacy in treating other thrombotic microangiopathies, there is insufficient data to support first-line treatment in CAPS. After the failure of first-line triple therapy in our patient, we planned for the use of rituximab as salvage therapy, but they rapidly deteriorated prior to initiation of treatment. Left or right ventricular assist devices have also emerged as an alternative treatment option for cases with severe heart failure, with varying success [7,8].
A multidisciplinary team conference was held, and it was determined that further medical and surgical care would be futile, given the extent of organ necrosis. The patient was transitioned to comfort care and eventually succumbed to multiorgan failure.
Conclusion
This is a rare case demonstrating the rapid progression of CAPS, resulting in systemic thromboses and fatal myocardial dysfunction. While guideline-directed triple therapy has demonstrated improved survival in many patients, a large subset continues to fail treatment. Second-line treatment with monoclonal antibodies and less common therapies such as ventricular assist devices may be warranted in aggressive treatment of some patients with refractory and/or rapidly progressive disease.
Disclosures: The authors have nothing to disclose.
Funding: The authors have no funding to disclose.
References
- Cervera R, Rodríguez-Pintó I, Legault K, Erkan D. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Catastrophic Antiphospholipid Syndrome. Lupus. 2020; 29(12): 1594-1600. doi: 10.1177/0961203320951260.
- Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, et al. Catastrophic Antiphospholipid Syndrome (CAPS): Descriptive analysis of a series of 280 patients from the CAPS Registry. J Autoimmun. 2009; 32(3-4): 240-5. doi: 10.1016/j.jaut.2009.02.008. Epub 2009 Mar 26.
- Girish B, Gainder S, Saha SC, Krishnappa D. Rare Presentation of Catastrophic Antiphospholipid Syndrome with Myocarditis in Post-partum Period: Case Report and Review of Literature. J Obstet Gynaecol India. 2018; 68(1): 70-72. doi: 10.1007/s13224-017-0974-7.
- Berman H, Rodríguez-Pintó I, Cervera R, Morel N, Costedoat-Chalumeau N, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab. Autoimmun Rev. 2013; 12(11): 1085-90. doi: 10.1016/j.autrev.2013.05.004.
- Unlu O, Erkan D. Catastrophic Antiphospholipid Syndrome: Candidate Therapies for a Potentially Lethal Disease. Annu Rev Med. 2017; 68: 287-296. doi: 10.1146/annurev-med-042915-102529.
- Legault K, Schunemann H, Hillis C, Yeung C, Akl EA, et al. McMaster RARE-Bestpractices clinical practice guideline on diagnosis and management of the catastrophic antiphospholipid syndrome. J Thromb Haemost. 2018; 16(8): 1656-1664. doi: 10.1111/jth.14192.
- Tarzia V, Tessari C, Fabozzo A, Cavalli C, Pagnin C, et al. Antiphospholipid antibody syndrome and LVAD: What are the chances? A case report and literature review. Int J Artif Organs. 2022; 45(2): 235-238. doi: 10.1177/0391398821996726. Epub 2021 Mar 18.
- Pólos M, Kovács A, Németh E, Merkely B. Acute thrombosis of the ascending aorta causing right ventricular failure: First manifestation of antiphospholipid syndrome. Eur J Cardiothorac Surg. 2019; 55(2): 371-373. doi: 10.1093/ejcts/ezy218.
Supplementary files
Video files: https://drive.google.com/drive/folders/1mwxr14QuHpUzFhIqKureqlvIND2rmIfP?usp=drive_link
Legends
Video 1: [2D Transthoracic Echocardiogram – Apical Four Chamber View without Contrast. Severe diffuse hypokinesis with an estimated EF of 10-15%, regional dyskinesia of the apical segment, and a mildly dilated left ventricle cavity.
Video 2: [2D Transthoracic Echocardiogram-Apical Four Chamber View with Contrast. Severe diffuse hypokinesis with an estimated EF of 10-15%, regional dyskinesia of the apical segment, and a mildly dilated left ventricle cavity.]
Video 3: Cardiac MRI - CINE Four Chamber View. Severely dilated left ventricle and reduced left ventricular systolic function, suggestive of dilated cardiomyopathy. Akinesis of mid-apical anterior, anterolateral, anteroseptal wall segments, hypokinesis of mid-apical inferior, inferoseptal, inferolateral wall segments. Mild-to-moderate pericardial effusion is also noted, supportive of myocarditis.
Video 4: Left Heart Catheterization - LAO Caudal View. Moderate to severe stenoses of the left anterior descending and left circumflex arteries.
Video 5: Left Heart Catheterization - RAO Cranial View. Distal disease evident in right coronary arteries.