
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Short Commentary - Open Access, Volume 5
Primary amenorrhoea in a case with homozygous T(14;21) in a family with consanguinity
Bani Bandana Ganguly1,2*; Nitin N Kadam1,2
1MGM Center for Genetic Research & Diagnosis, MGM New Bombay Hospital, Navi Mumbai, India.
2MGM Institute of Health Sciences, Navi Mumbai, India.
*Corresponding Author : Bani Bandana Ganguly
MGM Center for Genetic Research & Diagnosis, MGM New Bombay Hospital, Navi Mumbai, India.
Tel: 91-22-61526527 & 91-22-27824618;
Email: mgmgeneticlab@yahoo.com
Received : May 16, 2024
Accepted : Jun 10, 2024
Published : Jun 17, 2024
Archived : www.jcimcr.org
Copyright : © Ganguly BB (2024).
Abstract
Consanguineous marriage shares blood lineage and is known to cause genetic defect in progenies through transmission of detrimental genes from both parents. Autosomal recessive disorders are well known in this context. Transmission of chromosomal rearrangements through consanguinity is not a very common example. Rearrangements involving chromosome 21 poses additional risk of trisomy 21 or Down Syndrome (DS) in the offspring. We present a 18 year old girl, referred for karyotyping for primary amenorrhoea, and that detected homozygous 44, XX, 2xt(14;21) chromosomal pattern in her genome. Retrospectively parental consanguinity and presence of t(14;21) in her mother and paternal uncle confirmed inheritance of t(14;21) from both parents. Her brother and paternal cousin too were detected with the translocation, where the latter had trisomy 21 i.e. DS indicating higher risk of DS in progenies of the carriers of t(14;21). The novelty of the case is many-fold: involvement of 21 in translocation influences meiotic error; 2xt(14;21) caused primary amenorrhoea; consanguinity has resulted in inheritance of t(14;21) from both parents; complete absence of free 14 and 21 in a single individual; and application of conventional cytogenetics for detection of this condition at an affordable cost. Culture of consanguinity shall be abolished to protect next generation from the burden of genetic disorders.
Keywords: Consanguinity; Homozygous 44, XX, 2x(14;21); Primary amenorrhoea; Trisomy 21; Down syndrome.
Citation: Ganguly BB, Kadam NN. Primary amenorrhoea in a case with homozygous T(14; 21) in a family with consanguinity. J Clin Images Med Case Rep. 2024; 5(6): 3123
Case presentation
An 18 year old girl with history of primary amenorrhoea was referred for karyotyping. She was tall (5.5 ft) and appeared healthy with normal look. She was retrospectively detected having born to consanguineous parents. She did not have any other medical condition; thus, they declined to go for further investigation. Her parents were first cousins. She had one younger sibling alive and one elder brother, who expired at early age and the family could not describe the reason of his death. His karyotyping was not performed.
The karyotype of the present case appeared with 44 chromosomes with two copies of homozygous Robertsonian translocation involving 14 and 21 in each. She had two normal X chromosomes with 44, XX, 2xt(14;21) karyotypic pattern in all 25 cells analysed (Figure 1a). Based on this result, karyotyping was considered for her mother and the younger sibling, and that revealed 45, XX, t(14;21) (Figure 1b) and 45, XY, t(14;21) (Figure 1c) of the mother and brother respectively meaning they had heterozygous condition of the translocation. Father was not alive. One of the paternal cousins (born to paternal uncle) had congenital malformations with mongoloid facial features and karyotyping revealed 46, XY, +21, t(14;21) pattern with trisomy 21 and heterozygous t(14;21) in all 25 cells studied. Thus, he was diagnosed as a variant Down syndrome. Further karyotyping of the paternal uncle showed 45, XY, t(14;21) pattern. Consanguinity and carrier status of the paternal or maternal grandparents remained unknown.
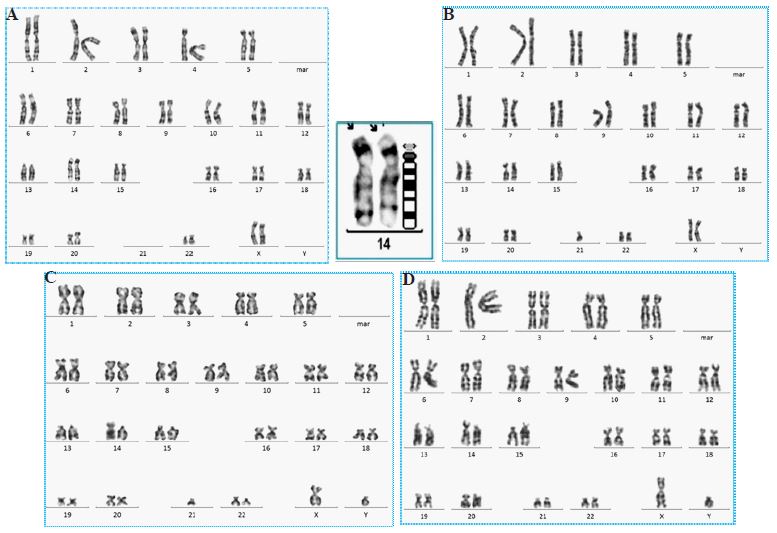

It is apparent from all these results that her father also had t(14;21). She had inherited the t(14;21) from both parents (44, XX, t(14;21) pat, t(14;21)mat) and her sibling received one copy from either of the parents. This is inheritance of genetic condition from parents (Figure 2). However, the girl was not lucky because she inherited parental abnormal genetic property from both parents, not economy.
Material and methods
For karyotyping, 5 ml of peripheral venous blood was collected in sodium heparin. Whole blood culture was performed in 5 ml RPMI medium supplemented with fetal bovine serum and phytohemagglutinin in replicate sets [1]. The cells were harvested following standard colchicine-hypotonic-fixative protocol, dropped onto chilled slides and air dried. Metaphase chromosomes were trypsinized and stained in Giemsa following standard GTG banding technique. A total of 25 well spread metaphases were analysed for each subject, and karyotypic classification was considered for 10 metaphases for each following ISCN nomenclature (ISCN 2020).
Discussion
Consanguinity is a cultural practice of some of the Indian communities, especially in minorities of tight religious faith [2,3]. The system is known to multiply the risk of hereditary condition of recessive mutations in future generations and affects births with genetic impairments. The autosomal recessive genetic conditions are expressed clinically in children born to consanguineous parents. Jews Genetic Diseases (JDG) of the Ashkenazi Jews population and many such racial recessive genetic disorders have been established through consanguineous marriages [1].
Robertosonian translocations have been reported as the frequently seen structural rearrangements with an epidemiologic prevalence of 1 in every 1000 newborns. These translocations arise from inter-chromosomal exchanges of the acrocentric chromosomes, which may occur at the centromere region, in the proximal short-arm satellite DNA, or within the Nucleolar-Organizing Region (NOR) of ‘D’ and ‘G’ group chromosomes. The inter-chromosomal rearrangements result in monocentric or dicentric chromosomes, depending on the site of the recombination event [4]. The configuration of the two acrocentric chromosomes changes to metacentric (D; D or G; G) or sub-metacentric chromosomes (D; G). In the present report, the translocation in all carriers became sub-metacentric due to fusion of 14 and 21. Thus, the fusion of two centromeres of the two acrocentric chromosomes results in either loss of centromere of one chromosome or centromere of both chromosomes could be retained as a dicentric chromosome. Nevertheless, the translocation results in loss of satellites of both acrocentric chromosomes [5]. Severity of clinical consequences might depend on the content of the centromere in Robertsonian translocations. The individuals carrying t(14;21) may not have any discernible clinical or phenotypic effect; however, they are a greater risk of experiencing meiotic non-disjunction, and thus, reproduces children with DS with Robertsonian translocation meaning these children carry both numerical and structural alterations.
The present case has cited a unique example of inheritance of chromosomal abnormality from both parents, who are first cousins and have shared blood lineage. In general, carriers of t(14;21) apparently have full genetic complement and are fertile, but with higher risk of pregnancies with trisomy 21 i.e. DS due to production of unbalanced gametes [4]. Seemingly, presence of t(14;21) in the paternal uncle might have influenced meiotic non-disjunction leading to trisomy 21, and that had resulted in DS in the paternal cousin of the propositus. The present case did not attain menarche, though she had two normal X chromosomes. Hence, she would most likely be infertile. Her younger brother carrying one copy of the translocation might not suffer from primary infertility, but might reproduce children with DS similar to the situation of the paternal uncle.
Homozygous Robertsonian translocations between 14 and 15, 13 and 14, and 14 and 21 have been reported in individuals born to consanguineous and heterozygous carriers [6,7]. A DS karyotype was reported with homozygous t(14;21) [8]. Prenatal diagnosis would be mandatory for heterozygous carriers, including her sibling, who is carrying a t (14;21), since that poses a risk of trisomy 21 in future generation. The proband, if attains menarche and is successful to conceive a pregnancy, in all normal consequences she will transmit one copy of the Robertsonian translocation to her offspring. However, since both of her 14s and 21s are translocated, a complex recombination can occur from crossing over during gametogenesis. Two copies of homozygous t(14;21) was reported in a fetus who was physically normal at birth and had a normal psychomotor development [9]. The present case was healthy and didn’t have any dysmorphic features; thus, it corroborates well with subjects with 44 chromosomes and with homozygous Robertsonian translocations, though ribosomal rDNA gene clusters, several long noncoding RNAs, and in the case of RTs involving chromosome 21, several mRNA encoding genes are perturbed [10]. Thus, since consanguinity shares blood lineage, it is detrimental for the next generation; hence, the practice shall be avoided.
The present study has employed conventional G-banding technique exclusively on peripheral blood lymphocytic chromosomes, and the technique was good enough for the present family study and detection of chromosomal rearrangements at pre- and post-natal age [11]. Previous publication with homozygous (14;21) translocation in a Down syndrome had considered Fluorescence In Situ Hybridization (FISH) and microsatellite polymorphism assay for confirmation of the origin of the two t (14;21) s [8]. Another study of homozygous t (14;15) involved FISH, array-comparative genomic hybridization (a-CGH), and telomere FISH on peripheral blood and spermatocytes of the hetero- and [7]. However, the two studies have detected parental non-consanguinity and segregation of the translocation in hetero- and homozygous carriers. It is needless to mention that conventional G-banding and pedigree analyses are sufficient to establish the inheritance and segregation. In all reports, conventional cytogenetics was the first technique employed to detect the presence of chromosomal rearrangements. Therefore, the present study didn’t require employment of other molecular technique for generating the information included in this report.
Acknowledgements: The author gratefully acknowledges the patient’s family for their participation and consent in reporting this study, and to Mahatma Gandhi Mission Trust for providing facilities for the investigation.
Funding: Nil.
References
- Ganguly BB, Kadam NN. Genetic Race: Prevalence of Diseases and Guidelines for Prevention. MGM J Med Sci. 2019; 6(1): 32-41. 10.5005/jp-journals-10036-1228.
- Acharya S, Sahoo H. Consanguineous Marriages in India: Prevalence and Determinants. Journal of Health Management. 2021; 23(4): 631-648. doi:10.1177/09720634211050458.
- Sharma SK, Kalam MA, Ghosh S, Roy S. Prevalence and determinants of consanguineous marriage and its types in India: evidence from the National Family Health Survey, 2015-2016. J Biosoc Sci. 2021; 53(4): 566-576. doi: 10.1017/S0021932020000383.
- Ganguly BB. Genetics and Neurobiology of Down syndrome. Elsevier, Academic Press, NY, USA. 2022. DOI: 10.1016/B978-0-323-90456-8.00013-2.
- Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, et al. Down syndrome. Nat Rev Dis Primers. 2020; 6(1): 9. doi: 10.1038/s41572-019-0143-7.
- O’Neil ID. Homozygosity for constitutional chromosomal rearrangements: A systematic review with reference to origin, ascertainment and phenotype. J Hum Genet. 2010; 55(9): 559-564.
- Song J, li X, Sun L, et al. A family with Robertsonian translocation: A potential mechanism of speciation in humans. Mol Cytogenet. 2016; 48(9). https://doi.org/10.1186/s13039-016-0255-7.
- Rajangam S, Michaelis RC, Velagaleti GV, Lincoln S, Hegde S, et al. Down syndrome with biparental inheritance of der (14q21q and maternally derived trisomy 21: Confirmation by fluorescent in situ hybridization and microsattelite polymorphism analysis. Am J Med Genet Part A. 1997; 70: 43-47.
- Dallapiccola B, Ferranti G, Altissimi D, Colloridi F, Paesano R. First-trimester prenatal diagnosis of homozygous (14;21) translocation in a fetus with 44 chromosomes. Prenat Diagn. 1989; 9: 555-558.
- Poot M, Hochstenbach R. Prevalence and Phenotypic Impact of Robertsonian Translocations. Mol Syndromol. 2021; 12: 1-11. DOI: 10.1159/000512676.
- Ganguly BB, Kadam NN. Prenatal diagnosis of fetus with short limbs caused by three abnormal chromosomes inherited from both parents. Int J Hum Genet. 2014; 14(2): 83-90.