
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Clinical Image - Open Access, Volume 5
Dystrophic Epidermolysis Bullosa (DEB): First looks
*Corresponding Author : Atul Jindal
Division of Pediatric Pulmonology and Critical Care, Department of Pediatrics, All India Institute of Medical Sciences, Raipur, Chhattisgarh, India.
Email: dratuljindal@gmail.com
Received : May 15, 2024
Accepted : Jun 14, 2024
Published : Jun 21, 2024
Archived : www.jcimcr.org
Copyright : © Jindal A (2024).
Citation: Jindal A. Dystrophic Epidermolysis Bullosa (DEB): First looks. J Clin Images Med Case Rep. 2024; 5(6): 3134
Description
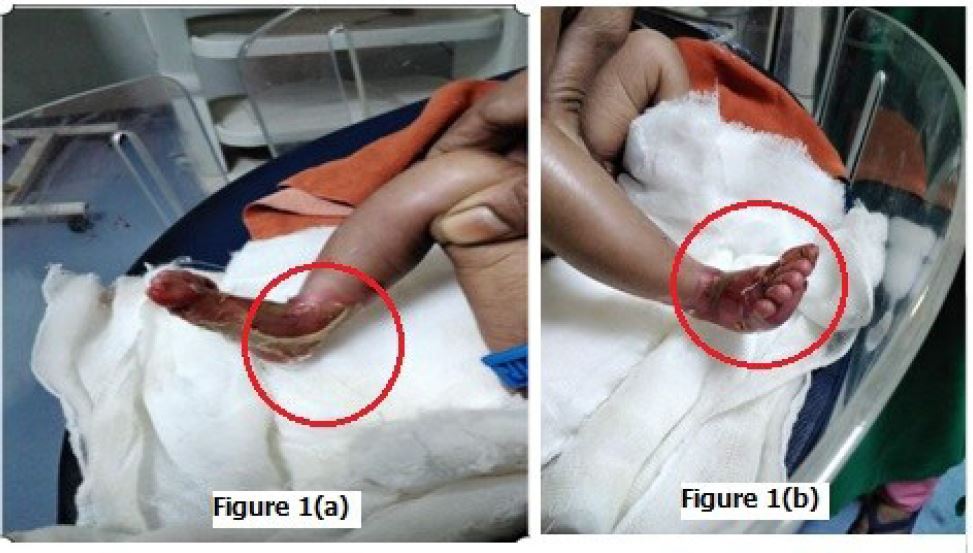
Dystrophic epidermolysis bullosa (DEB) is a severe genetic skin disorder caused by mutations in the collagen formation, resulting in extensive blistering, milia, and scarring. This condition is often accompanied by nail dystrophies, as well as changes in teeth and hair. DEB is commonly associated with severe deformities, leading to hand malformations resembling mittens, and an increased risk of squamous cell carcinoma. Newborns with DEB typically exhibit widespread blistering at birth, particularly on the ankles and neck, due to the forces applied during delivery [1]. This is a result of mutations in the COL7A1 gene, with at least 18 known mutations, and is inherited in a recessive manner. Dystrophic changes can also be observed in the nails. DEB is a debilitating childhood disease that causes easy trauma and painful recovery, often resulting in disfiguring scars [2]. The diagnosis is typically made through histopathology and immune-fluorescence testing, as there is no specific genetic test available. Unlike epidermolysis bullosa, DEB does not show immune-fluorescence due to antibody reactions.
Close differential diagnosis is pyoderma. This disease, however is characteristically different with presence of pustules, typically not presenting at birth, unlike DEB. Other differential is congenital herpes, but due to absence of lesions on the mother’s genital, no raised pro-inflammatory markers and no systemic involvement, this diagnosis can be ruled out. The presence of typical scarring and irreversible loss of nail is very typical of recessive DEB, with chances of other diseases unlikely. Attached (Figure 1(a) and 1(b)) show the typical findings in a neonate with recessive DEB [3].
There is an absence of a definitive management strategy for recessive DEB, thus various methods are utilized to address this debilitating disease. The primary approach focuses on increasing fibroblasts in the affected layer. This can be achieved through either the systemic injection of fibroblast stem cells or the local injection of genetically modified fibroblasts. Definitive management options include gene therapy, protein replacement, and cell-based approaches. Stem cell transplantation also holds potential. However, there is a lack of specialized centers in remote areas of India. In such circumstances, the only form of management available to patients is counseling for parents on wound care, prevention and early treatment of secondary infections, pain management, and occupational and physical therapy to address the inevitable deformities associated with this condition. Unfortunately, in economically disadvantaged and medically underserved regions, expectant treatment is the only solace that can be offered to these parents.
References
- Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. Journal of the American Academy of Dermatology. 2014; 70(6): 1103-26.
- Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner‐Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A, Marinkovich MP, Martinez AE. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. British Journal of Dermatology. 2020; 183(4): 614-27.
- Prost-Squarcioni CA, Caux F, Schmidt E, Jonkman MF, Vassileva S, Kim SC, Iranzo P, Daneshpazhooh M, Terra J, Bauer J, Fairley J. International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita. British Journal of Dermatology. 2018; 179(1): 30-41.
- Moravvej H, Abdollahimajd F, Naseh MH, Piravar Z, Abolhasani E, Mozafari N, Niknejad H. Cultured allogeneic fibroblast injection vs. fibroblasts cultured on amniotic membrane scaffold for dystrophic epidermolysis bullosa treatment. British Journal of Dermatology. 2018; 179(1): 72-9.