
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Short Report - Open Access, Volume 5
A rare case of Benign Recurrent Intrahepatic Cholestasis (BRIC) presenting as an unexplained cholestasis jaundice
Dhruv Madaan*; Raju H Badiger
J.N. Medical College, Nehru Nagar, Belagavi-590010, Karnataka, India.
*Corresponding Author : Dhruv Madaan
J.N. Medical College, Nehru Nagar, Belagavi-590010, Karnataka, India.
Email: dhruvmadaan75@gmail.com
Received : May 16, 2024
Accepted : Jun 20, 2024
Published : Jun 27, 2024
Archived : www.jcimcr.org
Copyright : © Madaan D (2024).
Citation: Madaan D, Badiger RH. A rare case of Benign Recurrent Intrahepatic Cholestasis (Bric) presenting as an unexplained cholestasis jaundice. J Clin Images Med Case Rep. 2024; 5(6): 3143.
Introduction
Benign Recurrent Intrahepatic Cholestasis (BRIC) is a very rare autosomal recessive disorder characterized by episodes of recurrent cholestatic jaundice manifested as pruritus, anorexia, fatigue, steatorrhea followed by complete resolution. Inheritance follows an autosomal recessive pattern with mutation in both alleles of ATP8B1 (BRIC1) or ABCB11 (BRIC2). It causes cholestasis by impairing the function of the bile salt export pump. The index episode usually occurs in the first two decades of life. Episodes can be spontaneous or triggered by infections or pregnancy and can last from weeks to months. Recognition of BRIC is important as it can lead to delayed or no diagnosis, also it is under-recognized and challenging. Diagnosis is based on a compatible clinical presentation, laboratory parameters and histology with exclusion of other causes of cholestasis.
Despite being recurrent, BRIC does not progress to advanced liver disease. We intend to report this case due to the rarity of this disease in India.
Case presentation
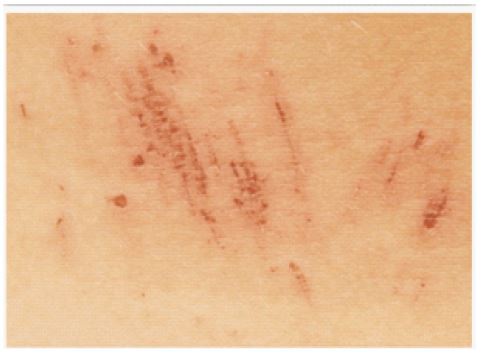
A 23-year-old female patient was admitted with an 8-week history of jaundice. Jaundice was accompanied by symptoms of pruritus (Figure 1), high coloured urine and clay coloured stools, which started during the last week of her third trimester of her third pregnancy and presenting even after the delivery. There was no history of accompanying pain abdomen, drug exposure, fever, nausea and vomiting, diarrhea. She had 3 similar episodes in the past at the ages of 13, 18 and 21 years.
The first episode had lasted for about 2 weeks, second episode for 10 weeks after her first pregnancy and third episode lasted for 6 weeks after her second pregnancy. All the episodes subsided spontaneously without any treatment. She reported history of similar episodes in her elder sister as well. She had no history of viral hepatitis, food or drug allergies, alcohol and tobacco abuse. On examination, she is conscious and coherent, poorly built and nourished with a BMI of 17.5. Pulse rate-72/min, blood pressure 110/80 mmHg in the right upper limb in the supine position. Icterus present and scratch marks are present all over the body. No Kayser-Fleischer ring, cyanosis, clubbing, pedal edema or lymphadenopathy. No signs of liver cell failure. No organomegaly or ascites on palpation of abdomen and bowel sounds heard. The respiratory, cardiovascular and central nervous system was unremarkable.
Investigations
Viral markers for hepatitis A, B, C, E; HIV 1 & 2; EBV and CMV were negative. Workups for autoimmune hepatitis and Wilson’s disease were negative. Antinuclear, anti-smooth muscle, anti- mitochondrial, anti-LKM1 antibodies are negative. Ultrasonography showed liver span of 11.5 cm with uniform echo-pattern with no evidence of local or diffuse pathology and maintained intra and extrahepatic biliary duct. Upper gastrointestinal scopy showed normal mucosa with varices.
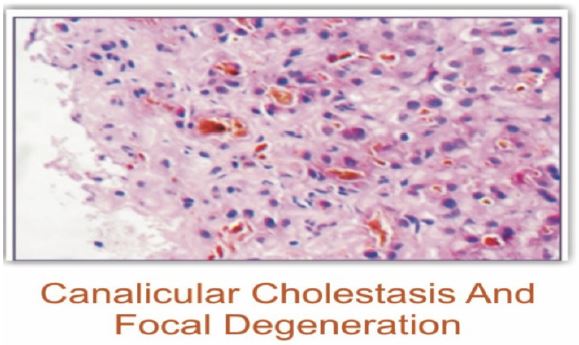
Magnetic resonance cholangiopancreatography showed normal intrahepatic and extrahepatic biliary ducts with no evidence of stone in the common bile duct. Liver biopsy showed a maintained lobular architecture comprised of hepatocytes showing scattered intrahepatic cholestasis and focal canalicular inflammation, bile canaliculi showing prominent intracanalicular cholestasis comprised dark brown coarse bile pigments, portal tracts show minimal lymphocytic infiltrates and no portal fibrosis, significant inflammation or nodule formation.
| Hb | Rbc | Platelet | TLC | Urea | Creat | Sodium | Potassium | INR | Amylase/lipase |
|---|---|---|---|---|---|---|---|---|---|
| 14.2 | 5.21 | 278 | 8.7 | 26 | 0.76 | 135 | 3.90 | 0.91 | 28/23 |
| Total bilirubin | Direct bilirubin | ALP | GGT | ALT | AST | |
|---|---|---|---|---|---|---|
| DAY 1 | 8.2 | 6.1 | 388 | 15.2 | 415 | 205 |
| DAY 7 | 12.4 | 9.8 | 421 | 14.8 | 354 | 106 |
| DAY 14 | 14.6 | 12.2 | 510 | 13.5 | 156 | 88 |
| DAY 21 | 18.0 | 16.5 | 615 | 12 | 80 | 60 |
| DAY 30 | 17.4 | 15.2 | 500 | 11.6 | 48 | 32 |
| DAY 45 | 12.24 | 9.6 | 480 | 10.2 | 38 | 22 |
| DAY 60 | 7.2 | 5.8 | 350 | 9.6 | 36 | 20 |
| DAY 90 | 1.6 | 0.8 | 86 | 9 | 28 | 15 |
Summary table of investigations
| Test/ Investigation | Results |
|---|---|
| Viral Markers | Negative for hepatitis A, B, C, E; HIV 1 & 2; EBV and CMV |
| Autoimmune Hepatitis Workups | Negative for autoimmune hepatitis and Wilson’s disease |
| Antibody Tests | Negative for Antinuclear, anti-smooth muscle, anti-mitochondrial, anti-LKM1 antibodies |
| Ultrasonography | Liver span of 11.5 cm with uniform echo-pattern; no local or diffuse pathology; maintained intra and extrahepatic biliary duct |
| Upper Gastrointestinal Scopy | Normal mucosa, no varices |
| Magnetic Resonance Cholangiopancre atography (MRCP) | Normal intrahepatic and extrahepatic biliary ducts, no evidence of stone in the common bile duct |
| Liver Biopsy | Maintained lobular architecture; scattered intrahepatic cholestasis; focal canalicular inflammation; dark brown coarse bile pigments in bile canaliculi; minimal lymphocytic infiltrates in portal tracts; no portal fibrosis, significant inflammation or nodule formation.
|
Diagnostic criteria for BRIC
At least two attacks of jaundice separated by a symptom-free interval lasting several months to years.
Laboratory values consistent with intrahepatic cholestasis - GGT either normal or only minimally elevated.
Severe pruritus secondary to cholestasis.
Liver histology demonstrating centrilobular cholestasis.
Normal intra- and extrahepatic bile ducts by cholangiography.
Absence of factors known to be associated with cholestasis.
Discussion
BRIC (Benign Recurrent Intrahepatic Cholestasis), first described in 1959, has since been reported to occur worldwide. Till date very few cases of BRIC have been reported, which may be due to the rarity of the condition compounded by under-recognition by clinicians. Inheritance follows an autosomal recessive pattern with mutations in both alleles of ATP8B1 (BRIC1) or ABCB11 (BRIC2). Both mutations cause cholestasis by impairing the function of the Bile Salt Export Pump (BSEP), which actively transports bile into canaliculi. Despite being a genetic disease, most cases are sporadic. The first episode usually occurs in the first two decades of life. Episodes can last from weeks to months and can be varying severity. Diagnosis in our case was based on the past episodes of jaundice, presenting clinical features, laboratory parameters and liver biopsy findings as proposed by Luketic and Shiffman for the diagnosis of BRIC. This is a self-limiting disease with no residual damage and treatment is aimed at relieving symptoms and shortening of episodes. High dose fat-soluble vitamins should be supplemented to prevent deficiency during prolonged episodes. Bile acid sequestrants and short-term use of Ursodeoxycholic acid may reduce pruritus but not the duration of episodes. Rifampicin, plasmapheresis have shown to relieve symptoms and shorten episodes. The effectiveness of Rifampicin is attributed to its enzyme-inducing effects. Endoscopic nasobiliary drainage is an effective treatment option in patients refractory to standard therapy. In between episodes, patients remain totally devoid of symptoms for periods ranging from months to years. Triggers include stress, pregnancy, respiratory and gastrointestinal infections.
Conclusion
Benign recurrent intrahepatic cholestasis should be considered in the differential diagnosis of recurrent intrahepatic cholestasis in young aged patients without signs of liver cell failure. This is a self-limited cholestatic disorder with a good prognosis. Early diagnosis and confirmation with liver biopsy help to prevent other expensive investigations. Patients can be managed symptomatically and followed up.
References
- Summer skill WH, Walshe JM. Benign recurrent intrahepatic cholestatic jaundice. Lancet. 1959; 2: 686-90.
- Ellen RL. Wilson syndrome of benign recurrent cholestasis. Am J Med. 1963; 35: 249-56.
- Samal SC, Kalyour B. Subramanyam DK, Sthanakar RR, Ramakant C. Benign Recurrent Intrahepatic Cholestasis. J Assoc Physicians India. 1996; 44: 562-70.