
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Efficacy of autologous stem cell transplantation for crystalline kappa light chain proximal tubulopathy early recurrence after kidney transplantation
Diogo Francisco1*; Ana Cristina Martins1; Pierre Isnard2; Ilyass Zouhry2; Rebecca Sberro-Soussan3; Laurent Frenzel4; Marie-Alexandra Alyanakian5; Jean-Michel Goujon5; Guy Touchard6; Jean Paul Duong-Van-Huyen2; Marion Rabant2
1Nephrology Department, Santa Cruz Hospital, Lisboa, Portugal.
2Department of Pathology, Necker Hospital University Paris Descartes, Paris, France.
3Kidney Transplantation, Necker Hospital University Paris Descartes, Paris, France.
4Hematology Department, Necker Hospital University Paris Descartes, Paris, France.
5Immunology Department, Necker Hospital University Paris Descartes, Paris, France.
6Pathology Department, Poitiers University Hospital, Poitiers, France.
*Corresponding Author : Diogo Francisco
Nephrology Department, Santa Cruz Hospital, Lisboa, Portugal.
Email: diogofrbfrancisco@gmail.com
Received : Jun 12, 2024
Accepted : Jun 27, 2024
Published : Jul 04, 2024
Archived : www.jcimcr.org
Copyright : © Francisco D (2024).
Abstract
Monoclonal gammopathy of renal significance is defined as kidney injury due to underlying immunoglobulin-secreting plasma-cell or B-cell clone. Here, we report the case of a patient with incomplete Fanconi Syndrome diagnosed two months after Kidney Transplantation (KT), which revealed crystalline k-Light-Chain Proximal Tubulopathy (LCPT) on the kidney biopsy. Further investigation demonstrated 5% of dystrophic monoclonal plasma cells in the bone marrow. Clone-targeting chemotherapy followed by autologous stem cell transplantation were rapidly effective achieving very good partial response. Four subsequent biopsies performed up to 2 years post diagnosis showed no LCPT relapse. Hematological response was still maintained after 5 years. Monoclonal gammopathies of renal significance frequently relapse after transplant and there are currently no specific recommendations on treatment. ASCT following chemotherapy may be an efficient therapeutic option.
Keywords: Fanconi syndrome; Kidney transplantation; Monoclonal gammopathy; Hematopoietic stem cell transplantation.
Abbreviations: ASCT: Autologous Stem-Cell Transplantation; ESRD: End-Stage Renal Disease; FLC: Free Light Chain; FS: Fanconi Syndrome; KT: Kidney Transplantation; LCPT: Light Chain Proximal Tubulopathy; MGRS: Monoclonal Gammopathy of Renal Significance.
Citation: Francisco D, Martins AC, Isnard P, Zouhry I, Sberro-Soussan R, et al. Efficacy of autologous stem cell transplantation for crystalline kappa light chain proximal tubulopathy early recurrence after kidney transplantation. J Clin Images Med Case Rep. 2024; 5(7): 3154.
Introduction
Monoclonal Gammopathies of Renal Significance (MGRS) are defined as a B-cell or plasma-cell clonal disorders that do not fulfil the criteria for malignancy yet produce a monoclonal nephrotoxic immunoglobulin that can lead to kidney injury [1-3]. Despite an elevated rate of recurrence, MGRS is not a contra-indication for Kidney Transplantation (KT). Control of clonal immunoglobulin before KT is highly recommended, as is its close follow-up after treatment [2,4]. Here, we present a case of Fanconi Syndrome (FS) diagnosed early after KT that unveiled crystalline kappa Light Chain Proximal Tubulopathy (LCPT) as the most likely native kidney disease. Clone-targeted therapy with autologous stem cell transplantation led to resolution of the histologic signs of the disease and maintained hematological response.
Case report
A 57-year-old woman with presumed diabetic nephropathy developed end-stage renal disease 7 years after diagnosis. She had no other relevant medical history and no macro- or microvascular complications from diabetes. In November 2014 she underwent KT with a living-donor. She received induction therapy with basiliximab and maintenance immunosuppressive regimen including low dose tacrolimus, low dose everolimus and steroids. With an uneventful postoperative period, serum creatinine stabilized at 0.88 mg/dL. Two months after transplantation she developed acute rise in serum creatinine up to 2,15 mg/dL. Tacrolimus and everolimus blood through levels were within the normal range. No donor specific antibodies were identified. Renovascular Doppler ultrasonography was normal. Urinalysis showed an incomplete FS, with proteinuria of 1.75 g/g, hypomagnesemia, hypophosphatemia with increased fraction excretion of phosphorus (42%) and normoglycemic glycosuria and hypouricemia.
An allograft kidney biopsy was performed.
Light microscopy showed normal glomeruli and no signs of rejection. In the tubules, green droplets within the cytoplasm of proximal epithelial cells on Masson Trichrome were seen. Immunofluorescence and immunohistochemistry staining for Kappa LC was negative in glomeruli and strongly positive in proximal tubules, whereas staining for Lambda LC was negative. Electron microscopy revealed numerous crystalline rhomboid inclusions within the cytoplasm of proximal tubules, which stained for Kappa LC on immunogold (Figure 1). Final diagnosis was crystalline kappa LCPT.
Further investigations revealed urinary Kappa Free Light Chains (FLC) representing 24% of proteinuria. Hypogammaglobulinemia with an excess of k (502 mg/L) and no λ-FLC were found in the serum. The bone marrow aspirate revealed 5% of dystrophic monoclonal plasma cells. Monoclonal κ-gammopathy of renal significance was diagnosed. Moreover, retrospective examination of day-zero serum protein electrophoresis showed hypogammaglobulinemia at 5.9 g/dL with 2 microparaproteins, Kappa and IgG Kappa.
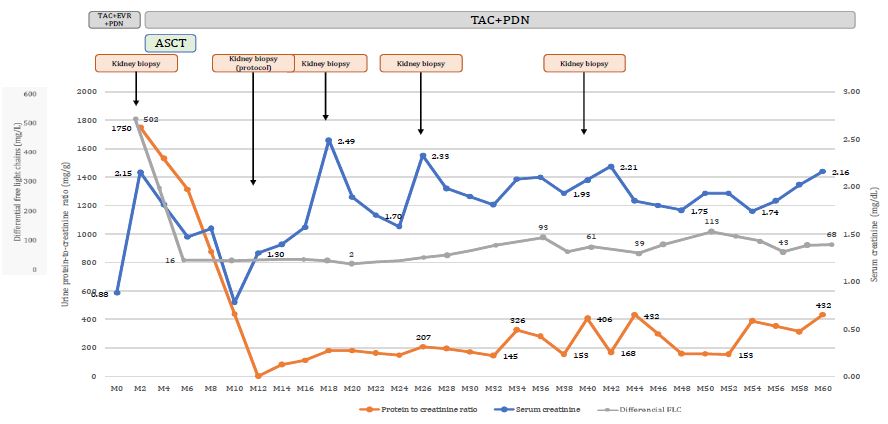
Everolimus was discontinued. After four courses of chemotherapy associating bortezomib, cyclophosphamide and dexa dexamethasone followed by high dose melphalan supported by Autologous Peripheral Blood Stem Cell Transplantation (ASCT), very good clinical and histological efficacy were achieved. Differential FLC (dFLC) decreased to 2 mg/dL immediately after treatment. Very Good Partial Response (VGPR) was maintained, despite fluctuations in dFLC after 30th month post-KT. In parallel, serum creatinine decreased and at one-year post-transplant measured glomerular filtration rate increased to 48 mL/min/1.73 m2 (Figure 2).
Early post-ASCT period was unremarkable. Despite fluctuation in both serum creatinine and dFLC, subsequent allograft biopsies performed at months 18, 26 and 40 post-KT detected neither LCPT recurrence, nor histologic signs of TMA (Figure 2).

Discussion
We describe a case of a 57-year-old KT recipient who developed incomplete Fanconi Syndrome two months after KT, unveiling crystalline κ-LCPT. Early recurrence in our case possibly reflects a high burden of monoclonal FLC at the time of KT and questions the initial diagnosis of diabetic nephropathy on the native kidney – which was not confirmed histologically.
Initial evaluation of MGRS includes assessment of the characteristics of the underlying clone, the type of nephropathy and its impact on renal function. As for other MGRS, the aim of the treatment of LCPT is to preserve renal function by targeting the underlying plasma cell or lymphoid cell clone that is producing the nephrotoxic M protein. Clone-targeting therapy is indicated if renal lesions are expected to be at least partially reversible and possible toxicities from treatment are acceptable according to each patient’s performance status. However, except for amyloidosis, there are currently no evidence-based recommendations for treatment of MGRS [1].
Anti-myeloma agents – such as bortezomib-based regimens – are used in plasmocytic disorders. Bortezomib has a non-renal metabolism and has proven efficacy even in patients with renal impairment with multiple myeloma [5]. In eligible patients, autologous stem cell transplantation appears to provide the best renal outcomes [6,7]. A decision to undergo ASCT should be made on a case-by-case according to risk-benefit assessment. In general, younger patients with good pulmonary and cardiac function and good performance status are eligible for ASCT1. In this case, presentation with rapid decrease in renal function early after KT in a young patient in whom the underlying clone might had led to ESRD made us recommend more aggressive treatment with chemotherapy and ASCT.
In a recent series from Heybeli et al, they report 3 cases of treatment-naive κ-LCPT patients who recurred at 25,68- and 423-days post-KT [8]. One patient did not receive treatment after the recurrence. A second patient achieved Very Good Partial Response (VGPR) with bortezomib-based therapy. Despite return to baseline renal function, 5-year protocol biopsy still detected LCPT. A third patient with early recurrence was treated with ASCT and achieved VGPR. However, the patient died of infectious complications 25 months post-KT. Another case of treatment-naive LCPT recurrence after KT was described by Angioi et al. [9] in whom a bortezomib-based therapy, led to VGPR.
Assessment of response to therapy relies both on renal – eGFR and proteinuria – and hematological parameters – serum FLC and urine and plasma electrophoresis and immunofixation.Absence of detectable monoclonal light chain defines complete remission but VGPR – defined as dFLC less than 40 mg/dL or at least a 90% reduction in the involved FLC – correlates with better renal outcomes [2,4,6,10,11]. In our patient, renal function stabilized at an eGFR of 33 mL/min/1.72 m2 with low grade proteinuria. Despite fluctuations in dFLC during follow up, no monoclonal FLC were detected and neither LCPT nor aspects of TMA were identified in the 4 subsequent biopsies that were performed up to 2 years after treatment and no further clone-targeting chemotherapy was administered.
There are currently no recommendations on immunosuppression modification strategies in patients with monoclonal gammopathies [11]. In our case, reduction of overall immunosuppression to a two-drug regimen was adopted with no subsequent rejection. There are no reports on higher rates of rejection or infection among patients who undergo ASCT or treatment with proteasome inhibitors for AL Amyloidosis. However, treatment with immunomodulatory drugs such as lenalidomide have been implicated with higher risk of rejection [11,12].
To summarize, we describe a case of a patient with ESRD who was diagnosed with crystalline κ-LCPT two months after KT. Treatment with chemotherapy and ASCT delivered favorable renal and hematologic response, up to 5 years after treatment and histological documentation up to 2 years, with no LCPT recurrence.
References
- K Amaador, H Peeters, MC Minnema, TQ Nguyen AD, JMI Vos, AJ Croockewit, NWCJ van de Donk, JFM Jacobs, JFM Wetzels, B Sprangers, AC Abrahams. Monoclonal gammopathy of renal significance (MGRS): histopathologic classification, diagnostic workup, and therapeutic options. The Netherlands Journal of Medicine. 2019.
- Leung N, Bridoux F, Nasr SH. Monoclonal Gammopathy of Renal Significance. N Engl J Med. May 20. 2021; 384(20): 1931-1941. doi:10.1056/NEJMra1810907.
- Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019; 15(1): 45-59. doi:10.1038/s41581-018-0077-4.
- Fermand JP, Bridoux F, Kyle RA, et al. How I treat monoclonal gammopathy of renal significance (MGRS). Blood. 21 2013; 122(22): 3583-90. doi:10.1182/blood-2013-05-495929.
- Sonneveld P, Schmidt-Wolf IG, van der Holt B, et al. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J Clin Oncol. 2012; 30(24): 2946-55. doi:10.1200/jco.2011.39.6820.
- Stokes MB, Valeri AM, Herlitz L, et al. Light Chain Proximal Tubulopathy: Clinical and Pathologic Characteristics in the Modern Treatment Era. J Am Soc Nephrol. 2016; 27(5): 1555-65. doi:10.1681/asn.2015020185.
- Gozzetti A, Guarnieri A, Zamagni E, et al. Monoclonal gammopathy of renal significance (MGRS): Real-world data on outcomes and prognostic factors. Am J Hematol. 2022; 97(7): 877-884. doi:10.1002/ajh.26566.
- Heybeli C, Alexander MP, Bentall AJ, et al. Kidney Transplantation in Patients With Monoclonal Gammopathy of Renal Significance (MGRS)-Associated Lesions: A Case Series. Am J Kidney Dis. 2022; 79(2): 202-216. doi: 10.1053/j.ajkd.2021.04.015.
- Angioi A, Amer H, Fervenza FC, Sethi S. Recurrent Light Chain Proximal Tubulopathy in a Kidney Allograft. Am J Kidney Dis. 2016; 68(3): 483-7. doi: 10.1053/j.ajkd.2016.04.021
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012; 30(36): 4541-9. doi:10.1200/jco.2011.37.7614.
- Angel-Korman A, Havasi A. Kidney Transplantation in Systemic Amyloidosis. Transplantation. 2020; 104(10): 2035-2047. doi:10.1097/tp.0000000000003170.
- Lum EL, Huang E, Bunnapradist S, Pham T, Danovitch G. Acute Kidney Allograft Rejection Precipitated by Lenalidomide Treatment for Multiple Myeloma. Am J Kidney Dis. 2017; 69(5): 701-704. doi: 10.1053/j.ajkd.2016.11.024.