
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
A female with 17-alpha-hydroxylase deficiency case report
Salimeh Dodangeh1; Akbar Soltani2; Zahra Hoseini Tavassol3; Shirin Hasani-Ranjbar3*
1Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
2Evidence Based Medicine Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
3Obesity and Eating Habits Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
*Corresponding Author : Shirin Hasani-Ranjbar
Obesity and Eating Habits Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
Tel: +98-21- 88220038;
Email: sh_hasani@tums.ac.ir &
shirinhasanir@yahoo.com
Received : Jul 15, 2024
Accepted : Jul 29, 2024
Published : Aug 05, 2024
Archived : www.jcimcr.org
Copyright : © Hasani-Ranjbar S (2024).
Abstract
17α-Hydroxylase Deficiency (17OHD), a rare form of congenital adrenal hyperplasia, results in impaired cortisol and sex steroid synthesis. Patients present with a female phenotype, high blood pressure, primary amenorrhea, lack of secondary sexual characteristics, and abnormal karyotypes (46XX or 46XY). In this article, we discuss a case of a 13-year-old female phenotype presented with the absence of secondary sexual characteristics, primary amenorrhea, absence of uterus, and normal blood pressure in the initial evaluation. A karyotype study suggested the presence of 46, XY chromosomal sex, and genetic analysis revealed a mutation in the CYP17A1 gene, thus confirming the diagnosis of 17α-hydroxylase deficiency.
Keywords: 17OHD; CAH; Amenorrhea.
Citation: Dodangeh S, Soltani A, Tavassol ZH, Hasani-Ranjbar S. A female with 17 alpha-hydroxylase deficiency case report. J Clin Images Med Case Rep. 2024; 5(8): 3192.
Introduction
Congenital Adrenal Hyperplasia (CAH) occurs due to defective steroid biosynthesis in adrenal and gonadal structures. Most CAH cases result from a deficiency of 21-hydroxylase or 11-beta-hydroxylase, accounting for 90% and 7% of cases, respectively [1,2]. 17 hydroxylase/17, 20-lyase deficiency is a rare form of CAH, affecting about 1% of all cases [2]. The essential role of the enzyme 17-alpha-hydroxylase is recognized as a part of the synthesis of cortisol and adrenal gland hormones and these hormones play a role in the adrenal steroid production [3]. 17-alpha-hydroxylase deficiency (17OHD), with autosomal recessive inheritance, leads to impaired cortisol and sex steroid synthesis, with a compensative increase of Adrenocorticotropic Hormone (ACTH), gonadotropins, corticosterone, and 11-Deoxycorticosterone (DOC) [4]. Severe form of disease present with female phenotype, hypertension and hypokalemia, primary amenorrhea, and absence of secondary sexual characteristics. Karyotype studying of these patients represents 46XX OR 46XY karyotype [5]. Elevated serum progesterone with or without elevated serum 17-alpha-hydroxyprogesterone and recurrent ovarian cysts are typical manifestations of partial 17OHD. Furthermore, hypokalemic hypertension may be absent in partial 17OHD [6]. In 46, XX patients, external genitalia on the neonatal exam and the internal one on ultrasound are normal. These patients may not have any complaints before pubertal age; however, they may develop hypertension or hypokalemia and a high level of gonadotropin (hypergonadotropic hypogonadism). Whenever sex hormone deficiencies become apparent in 46, XY patients, the typical presentation includes under-masculinization and ranges from phenotypic female to ambiguous or small male genitalia [5] (Figure 1). In this paper, we reported a normotensive female patient of 13-year-old 46, XY who had 17OHD with clinical presentations of primary amenorrhea and absence of secondary sexual characteristics and with laboratory tests and genetic mutations that confirmed 17OHD.
Case presentation
A 13-year-old girl was referred to the endocrinology clinic of Shariati Hospital with complaints of absence of secondary sexual characteristics. She had a medical history of two episodes of seizure at infancy and age 4 due to hypoglycemia, scoliosis, Kawasaki disease with clinical manifestation of fever, rash, and cervical lymphadenopathy at age 10, history of hypothyroidism at 9 years old, treated with levothyroxine and inguinal hernia surgery at infancy, unfortunately, the pathology report is unavailable as the patient was visited in our center at the age of 13. She was the second child of consanguineous parents. The patient has an older sister with menarche at age 12 and normal development of breasts and axillary hair. On physical examination, the patient was tall, with a height of 163 cm, weight of 48 kg, and Body Mass Index (BMI) of 18 kg/m2. Her blood pressure was normal with an appropriate pediatric cuff during the initial evaluation; however, it was slightly elevated at 124/89 mmHg (95th percentile) during subsequent visits and her heart rate was about 100 beats /minute. Breast growth and axillary hairs were absent and the external genitalia was female type but with no pubic hair. The biochemical and hormonal parameters of this case are described below. Serum cortisol measurements at 08:00 were decreased below the test threshold [0.6 μg/dL (6.24-18.0)] with a high plasma ACTH level of 182.7 pg/mL(7.2-63.3). The sex hormones Dehydroepiandrosterone Sulfate (DHEAS) and estradiol were low [3.0 μg/dL(57-395) and < 18.35 Pmol/L(45-854), respectively]; however, serum Luteinizing Hormone (LH) and Follicle-Stimulating Hormone (FSH) levels were elevated to 48 and 77 IU/L, respectively. The Thyroid Stimulating Hormone (TSH) was normal. Serum potassium was decreased to 3.2 mmol/L; Serum aldosterone 2.29 ng/dL (2.52-39) and plasma renin concentration was <1.8 μIU/mL; Serum concentrations of 17-OH (17 OHP) progesterone (0.33 ng/mL) was low, but progesterone (19.68 nmol/L) and Anti-mullerian hormone (17.260 ng/mL) were raised. The AMH and inhibin-B levels of our patient suggested that they had normal Sertoli-cell function. Response to the Human chorionic gonadotropin stimulation test was blunted. Clinical manifestations and biochemical results are summarized in Table 1.
By considering the possibility of disorders in sexual development, imaging and karyotype analysis were carried out. The karyotype was 46, XY, indicative of male sex. Abdominal MRI and ultrasound revealed no visible female internal genitalia, such as ovaries, uterus, and upper third of the vagina; however, two streaky structures suspected to be testicles were visualized in the pelvic cavity; the bone age was delayed to 8.5 years old. Genomic DNA was extracted from peripheral blood leukocytes and whole exome sequencing (WES-200X) was done. The analysis revealed a homozygous mutation on exon 1 of the CYP17A1 gene on chromosome 10q24.32.
The consent was obtained from the patient and her parents. About the choice of gender, we fully discussed it with the patient and her parents, and the patient fully understood the condition and identified herself as being of female gender. Estrogen replacement with estradiol valerate 0.5 milligrams per day was initiated to promote epiphyseal healing and maintain the female appearance. The patient was on levothyroxine sodium from 9 years old. Replacement therapy for preventing adrenal crises has not been initiated because some degrees of glucocorticoid activity of elevated levels of corticosterone and adrenal crises are rare in 17OHD.
Breast development started after initiating the administration of estrogen. As the psychosocial problem is a significant concern in the long term; regular monitoring of psychological condition and blood pressure and electrolyte has been recommended for this patient. For reconstructive surgery in our patient, vaginoplasty may be required to create typical female sexual anatomy. The data on gonadal malignancy risk for cases of 17OHD are limited and we did not recommend gonadectomy for our patient in this stage, however, in follow up it should be individualized to the patient and made by a specialized multidisciplinary team.
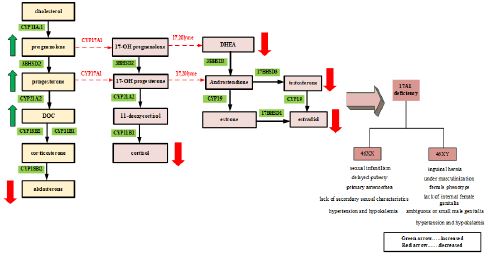
CYP11A1: 11α-hydroxylase; CYP 17A1: 17α-hydroxylase; CYP 21A2: 21α-hydroxylase; CYP11B1: 11β1-hydroxylase; CYP18B2:18β2-hydroxylase; CYP 19: 19α-hydroxylase (Aromatase); 3BHSD2:3β-hydroxysteroiddehydrogenase2; 17BHSD3:17β-hydroxysteroiddehydrogenase3; 17BHSD1:17β-hydroxysteroiddehydrogenase1; DOC: 11-deoxycorticosterone; CYP: Cytochrome P450.
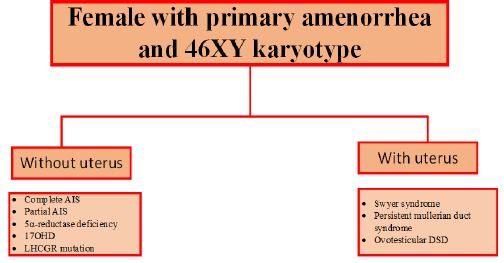
AIS: Androgen Insensitivity Syndrome; 17OHD: 17 alpha-hydroxylase deficiency; LHCGR mutation: luteinizing hormone/choriogonadotropin receptor mutation; DSD: Disorders of Sex Differentiation.
Table 1: The course of endoscopic treatments.
| Item | Result | Reference range | Item | Result | Reference range |
|---|---|---|---|---|---|
| Age (years) | 13 | AMH(ng/mL) | 17.260 | 0.9-9.5 | |
| Sexual sex | female | Inhibin-A(pg/mL) | 2.26 | 5.3-115 | |
| Height (cm) | 163 | Testosterone(ng/mL) | <0.02 | 0.03-0.68 | |
| Body weight (kg) | 48 | DHT(pg/mL) | 29 | 0-206 | |
| Blood pressure (mmHg) | 124/89 | TSH(μIU/mL) | 3.4 | 0.5-5 | |
| Karyotype | 46, XY | TT4(μg/dL) | 7.6 | 4.6-12.6 | |
| ACTH (pg/mL) | 182.7 | 7.2-63.3 | Sodium (mmol/L) | 144 | 135-145 |
| Cortisol(μg/dL) | 0.6 | 6.24-18.0 | Potassium (mmol/L) | 3.2 | 3.5-5 |
| 17OH- Progesterone(ng/mL) | 0.33 | 0.4-1.5 | Aldosterone(ng/dL) | 2.29 | 2.52-39 |
| Progesterone(nmol/L) | 19.68 | - | Plasma renin concentration(μIU/mL) | <1.8 | 5.3-99 |
| DHEAS (μg/dL) | 3 | 17-343 | |||
| Estradiol(pmol/L) | <18.35 | 45-85 | |||
| LH(mIU/ml) | 48 | 0.1-9.5 | |||
| FSH(mIU/ml) | 77 | 3.5-12.5 | |||
| Inhibin-B(pg/ml) | 76 | 22-85 |
ACTH: Adrenocorticotropic Hormone; AMH: Anti-Mullerian Hormone; DHEAS: Dehydroepiandrosteronesulfate; DHT: Dehydrotestosterone; FSH: Follicle Stimulating Hormone; LH: Luteinizing Hormone; TSH: Thyroid Stimulating Hormone; TT4: Total Thyroxine (T4).
Discussion
17-alpha-hydroxylase is encoded by the CYP17A1 gene, located on chromosome 10q24.3. Persons with a form of the disease account for about 1% of all CAH cases [7]. Severe form of 17OHD in 46, XY persons typically represent female phenotype accompanied by primary amenorrhea, absence of secondary sexual characteristics (absent axillary and pubic hair), female external genitalia with a blind vaginal pouch, and variable stage of hypertension usually diagnosed at adolescence. The testes may be discovered in the intra-abdominal or inguinal region or the labioscrotal folds. Partial form of 17OHD most frequently manifests in 46, XY infants with ambiguous genitalia or severe hypospadias with or without hypertension [8]. In our patient, the absence of significant hypertension, complete lack of virilization, and hypokalemia suggest a partial 17OHD. Testes produce normal levels of Anti-Mullerian Hormone (AMH), resulting in the absence of Mullerian structures (fallopian tubes, uterus and upper third of vagina). The development of external female genitalia is the consequence of decreased testosterone production. 17OHD blocks synthesis pathways of cortisol and sexual hormones and induces the mineralocorticoid pathway synthesis resulting in a decrease of 17-OHpregnolone, 17-OHP, 11-deoxycortisol, cortisol, DHEA-S, androstenedione, and testosterone. As a result of increased levels of DOC, sodium and fluid retention and loss of potassium and consequently hypertension, are common features in these patients. Despite decreased levels of cortisone, 17OHD patients rarely present with adrenal crises because of some degrees of glucocorticoid activity of elevated levels of corticosterone, preventing adrenal crises [9]. Most of these patients show low aldosterone levels due to elevated levels of DOC, resulting in suppression of the renin-angiotensin system [8], this was also in our case. We were unable to perform plasma levels of DOC. Our patient presented with female-type external genitalia, primary amenorrhea, absence of secondary sexual characteristics, partially elevated blood pressure, absence of Mullerian structures, and 46, XY karyotype which was studied based on this differential diagnosis (Figure 2): Androgen insensitivity syndrome, 5a-reductase deficiency, 17OHD and Leydig cell hypoplasia due to Luteinizing Hormone/Choriogonadotropin Receptor (LHCGR) mutation.
Diagnosis of 17α-hydroxylase deficiency was considered since none of those mentioned above except 17OHD will present with hypokalemia and partially elevated blood pressure in an individual with 46, XY Disorders of Sex Differentiation (DSD). Hypertension and, hypokalemia are the main features of 17OHD, because of infant kidney insensitivity to the mineralocorticoid effect of excess levels of DOC, this effect does not occur in infancy [8]. Hypertension may or may not be present in partial forms of combined 17α-hydroxylase/17, 20-lyase deficiency [10]. Another reason for missed diagnosis of elevated blood pressure is the unmeasurement of blood pressure ever young children with 17OHD. Such children are usually diagnosed during adolescence, because of delayed puberty, hypertension, and hypokalemia. The blood pressure of the patient was normal with an appropriate pediatric cuff during the initial evaluation; however, it was slightly elevated, during subsequent visits, indicating a partial deficiency of 17-alpha-hydroxylase, which is very rare. Therefore, children may not be recognized until puberty. Although this patient presents with normal blood pressure, spontaneous hypokalemia, and, suppressed plasma renin concentration strongly indicates the elevation of corticosterone or DOC. Decreased DHEA-S results in decreased testosterone excluding androgen insensitivity syndrome and 5a-reductase deficiency. Given the 46, XY karyotype, two streaky structures on pelvic MRI and low testosterone and high AMH and inhibin-B levels which indicate normal Sertoli cell function, as well as 17OHD, Leydig cell hypoplasia due to LHCGR mutation was considered before genetic testing. Therefore, Human Chorionic Gonadotropin (HCG) stimulation test was carried out. In this case, abnormal results of the HCG stimulation test, with no increase in testosterone levels consistent with defective androgen biosynthesis in 17OHD; however, Leydig cell hypoplasia due to LHCGR mutation cannot be ruled out. Our patient had scoliosis; however, the association between 17OHD and scoliosis was not identified. Birth defects likely affect the development of the spine bones due to estrogen and androgen deficiency, contributing to the onset of scoliosis. Seizures resulting from hypoglycemia in infants with glucocorticoid deficiency due to adrenal insufficiency are justifiable. No association between hypothyroidism and Kawasaki disease with 17OHD.
We know that despite the classification of 17OHD in the group of cases with a uterus in the categorization of the causes of primary amenorrhea, our patient had a 17-alpha-hydroxylase deficiency without a uterus, making this case notable due to the rarity of such occurrences.
Laboratory evaluations are essential to the determination of diagnosis. Based on clinical and available biochemical findings supported the diagnosis of 17OHD. The genetic study of this patient confirmed the diagnosis of 17OHD. Genetic testing is essential for the established diagnosis of 17OHD due to the presence of various clinical and biochemical findings.
Conclusion
Partial 17OHD is very rare and most patients were asymptomatic until puberty.
It is crucial to consider this diagnosis when a girl exhibits the absence or incomplete development of secondary sexual characteristics, along with different degrees of hypertension and hypokalemia or lack of virilization in 46, XY individuals.
The normal blood pressure should not rule it out.
Individuals may develop hypertension during the subsequent visits, indicating the importance of screening blood pressure, and potassium and regular monitoring for these individuals.
Declarations
Acknowledgments: We appreciate the patient and his family’s cooperation in this case report.
Contributors: SD wrote the manuscript under the direction of SH, AK. Data collection was done by SD and ZTH. SH and AK managed the patient and finalized the manuscript. All authors reviewed, edited, and approved the final manuscript.
Disclosures: The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest.
Informed patient consent for publication: All procedures performed in the research involving patients were by the ethical standards of the Tehran University of Medical Sciences and with the Helsinki Declaration. Informed consent was obtained from the patient and her parents for publication of this case report.
References
- Merke DP, Bornstein SR. Congenital adrenal hyperplasia. The Lancet. 2005; 365(9477): 2125-36.
- Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best practice & research clinical endocrinology & metabolism. 2009; 23(2): 181-92.
- Wang M, Wang H, Zhao H, Li L, Liu M, et al. Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese Han population. Clinical Hypertension. 2019; 25: 1-9.
- Miller WL. Mechanisms in endocrinology: Rare defects in adrenal steroidogenesis. European journal of endocrinology. 2018; 179(3): R125-R41.
- Chormanski D, Muzio MR. C 17 hydroxylase deficiency. 2019.
- Tian Q, Zhang Y, Lu Z. Partial 17-alpha-hydroxylase/17, 20-lyase deficiency-clinical report of five Chinese 46, XX cases. Gynecological Endocrinology. 2008; 24(7): 362-7.
- Maheshwari M, Arya S, Lila AR, Sarathi V, Barnabas R, et al. 17α-Hydroxylase/17, 20-lyase deficiency in 46, XY: our experience and review of literature. Journal of the Endocrine Society. 2022; 6(3): bvac011.
- Auchus RJ. Steroid 17-hydroxylase and 17, 20-lyase deficiencies, genetic and pharmacologic. The Journal of steroid biochemistry and molecular biology. 2017; 165: 71-8.
- Kim SM, Rhee JH. A case of 17-alpha-hydroxylase deficiency. Clinical and experimental reproductive medicine. 2015; 42(2): 72.
- Marsh CA, Auchus RJ. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17, 20-lyase deficiency and isolated 17, 20-lyase deficiency. Fertility and Sterility. 2014; 101(2): 317-22.