
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Pericardial lesion onset: A case of adult Erdheim-Chester disease and langerhans cell histiocytosis overlap syndrome
Leng Huang; Ling Qin*
Department of Respiratory Medicine, Xiangya Hospital, Central South University, Changsha 410008, Hunan, China.
*Corresponding Author : Ling Qin
Department of Respiratory Medicine, Xiangya Hospital, Central South University, Hunan, China.
Email: qlmelody@csu.edu.cn
Received : Jun 21, 2024
Accepted : Aug 21, 2024
Published : Aug 28, 2024
Archived : www.jcimcr.org
Copyright : © Qin L (2024).
Abstract
Erdheim-Chester Disease (ECD) is an uncommon type of non-Langerhans cell histiocytosis, distinguished by the presence of lipid-filled frothy macrophages infiltrating various tissues. Langerhans Cell Histiocytosis (LCH) is characterized by infiltration of Langerhans cells, which accumulate in various tissues and can cause a range of symptoms Both ECD and LCH can cause severe systemic disorder, and the overlap of these two diseases is rarely reported. We report a 49-year-old woman with an overlap syndrome of ECD and LCH. The patient developed a 13-month history of breath shortness and edema of both limbs. Computed tomography showed lesions in various systems, and gene testing identified a BRAF V600E mutation. The immunohistochemical analysis allowed the final diagnosis of ECD and LCH overlap syndrome. This is the first reported case of adult ECD and LCH overlap syndrome with pericardial lesion. Our case presents the successful diagnosis of ECD and LCH overlap syndrome with pericardial lesion, and the molecular mechanism of this overlap syndrome is also discussed.
Keywords: Histiocytosis; Erdheim-chester disease; Langerhans cell histiocytosis; Medullary tumor; Overlap syndrome.
Citation: Huang L, Qin L. Pericardial lesion onset: A case of adult Erdheim-Chester disease and langerhans cell histiocytosis overlap syndrome. J Clin Images Med Case Rep. 2024; 5(8): 3227.
Introduction
Histiocytosis is a rare disease with excessive production of specialized white blood cells, which can cause lesions in multiple organs. The “L” group of histiocytosis includes Erdheim-Chester Disease (ECD) and Langerhans Cell Histiocytosis (LCH), and they share similar clinical features [1]. ECD is a systemic non-Langerhans cell histiocytosis, characterized by an infiltration of lipid-laden macrophages and multinucleated giant cells [2]. It potentially impairs the heart, lungs, kidneys, and peritoneum. ECD is frequently observed in middle-aged adults, and it is easily misdiagnosed because of its atypical symptoms [3]. LCH originates from a clonal proliferation of Langerhans cells [4], with the potential to develop lesions in multiple organs, most frequently bone and skin [5]. Different from ECD, LCH affects children in most cases.
Both ECD and LCH are rare independent systemic disorders that can cause multiple organ impairment [6]. The prognosis of these two diseases is heterogeneous depending on risk factors and clinical symptoms. The morphological and immunohistochemical evaluations of LCH and ECD are different. ECD is characterized by the infiltration of tissues by CD68+/CD1a- foamy histiocytes [7], while LCH is defined by the accumulation of CD68+/CD1a+ histiocytes. In the genetic testing, the BRAF V600E mutation may serve as an early driver mutation in both diseases. In rare circumstances, the two diseases coexist in the same patient, with less than reported 100 cases up to date. Since multiple systems are involved, the diagnosis and treatment of such patients are extremely difficult [8]. In this report, we present an adult ECD and LCH overlap syndrome with pericardial lesion and atypical respiratory symptoms.
Case report
A 49-year-old woman visited the department of respiratory medicine because of a 13-month history of aggravating breath shortness and both lower limbs edema. She did not report cough, expectoration, fever, night sweats, weight loss, hemoptysis, or syncope. Five months earlier, her Interferon-γ–Release Assay (IGRA) for Mycobacterium tuberculosis showed a positive result. She was suspected to have tuberculous pericarditis and began diagnostic anti-tuberculosis therapy (rifampicin 0.45 g Qd + isoniazid 0.3 g Qd + ethambutol 0.75 g Qd + pyrazinamide 05 g Tid). During the following 4 months of treatment, she underwent three pericardial effusion drains. During this period, her symptoms worsened, and she came to hospital for diagnosis.
On admission, the patient’s blood pressure was 108/70 mmHg, her heart sounds were muffled and distant, the heart boundary expanded to the lower left, and both lower limbs were severely swollen. Laboratory tests showed a raised neutrophilia of 9.6×10⁹ per L, a raised C-reactive protein concentration (31.5 mg/L). IGRA for Mycobacterium tuberculosis was positive. Infectious diseases including HIV, syphilis, and hepatitis were excluded. Autoimmune diseases including systemic autoimmune diseases, IgG4-related diseases, and vasculitis were excluded. A transthoracic echocardiogram showed a severe pericardial effusion with 4 mm of the parietal pericardium and 5 mm of the visceral pericardium, and CT of the heart also showed extensive pericardial effusion (Figure 1A). A pericardiocentesis drained 1500 mL of pale-yellow fluid which suggested a transudate.
We continue to give anti-tuberculosis treatment and moxifloxacin (0.4 g Qd) following her previous treatment. However, PCR of Mycobacterium tuberculosis was negative in the pericardial effusion. During hospitalization, she often reported bone pain, which she often underwent in the past three years. We also noticed she had sparse pubic hair and symptoms of polydipsia and polyuria. Further laboratory tests show a normal blood osmotic pressure (300.0 mosm/kg.H2O) but with a low urine osmotic pressure (162.0 mosm/kg.H2O). After injection of desmopressin (5U), the patient’s urine osmotic pressure significantly increased to 477 mosm/kg.H2O, which indicated central diabetes insipidus. An abdominal CT showed multiple enlarged perihepatic lymph nodes, bilateral dilatation of the renal pelvises, and retroperitoneal fibrosis—known as hairy kidneys, (Figure 1B,C,D). MRI of the brain showed tuberous thickening of the pituitary stalk which indicated ECD of the brain (Figure 1E and F). There were multiple high-uptake lesions in the skull, vertebras, and bilateral ribs. A bone X-ray showed multiple long-bone osteosclerosis (Figure 1G and H). A whole-body 99mTc-MDP bone scintigraphy showed the “hot knee” sign: symmetrically increased uptake in the bilateral femur, tibia, and feet (Figure 1I).
The patient had thickened pituitary stalk, and enlarged lymph nodes, which highly suggested Langerhans Cell Histiocytosis (LCH). A perihepatic lymph node specimen provided evidence with infiltration of Langerhans histiocytes (Figure 1J): immunohistochemical analysis was positive for CD68, CD1a, S-100, and Langerin; genetic testing showed a mutation of BRAF V600E. However, Langerhans histiocytosis rarely presents pericardial lesion, “hairy kidney”, and “hot knee” sign, as these are consistent with the symptoms of Erdheim-Chester Disease (ECD). Considering the possibility of LCH and ECD overlap, we sent the patient’s pericardial effusion for pathology several times, which provided infiltration of non-Langerhans histiocytes (Figure 1K): immuno-histochemical analysis was positive for CD68, CD163, and negative for CD1a, S-100, and Langerin. These findings were consistent with the diagnosis of ECD and LDH overlap syndrome. Given the patient’s instable status, she was considered a poor candidate for immunotherapy. The patient’s symptom subsided after several pericardial effusion drains and supportive treatment. Then she continued with conservative treatment and returned to local hospital. Follow-up visits have shown clinical stability without any acute issues.
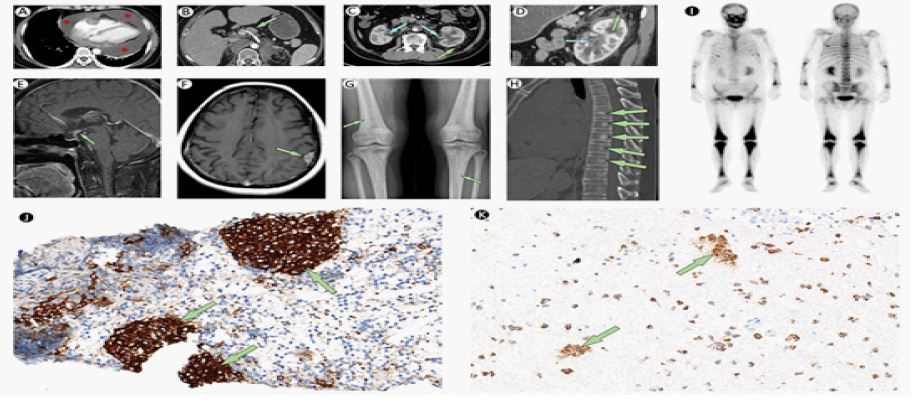
CT of the abdomen shows multiple enlarged perihepatic lymph nodes (green arrow; B), thickened renal pelvis walls (blue arrows; C,D) and hairy kidneys (green arrows; C,D).
MRI of the brain shows tuberous thickening of the pituitary stalk (green arrow; E) and Left parietal lobe enhancement focus (green arrow; F). X-ray of bone shows multiple long-bone osteosclerosis (green arrows; G).
CT of spine shows multiple bone sclerosis (green arrows; H).
The technetium-99m (99Tc) bones scintigraphy showed increased activity in many bones (I).
A lymph node puncture specimen shows Langerhans histiocyte infiltration (green arrows). Original magnification × 200 (J).
A pericardial effusion specimen shows Langerhans histiocyte infiltration (green arrow). Original magnification × 200 (K).
Discussion
ECD features the overgrowth of histiocytes [9], which invade multiple body systems and cause severe damage [10]. Diabetes insipidus is often considered as the first symptom of ECD, and the cardiovascular involvement is not rare [11]. ECD and LDH overlap syndrome is a rare reported multisystem disorder in adults. Nearly 15% of ECD patients also have LCH lesions, but as the incidence of ECD is low, the overlap syndrome of both diseases is exceptionally rare [7]. Since the diagnosis of ECD never precedes LCH, and both diseases have similar clinical complications, ECD can be easily overlooked in LCH patients. The reported cases of the overlap syndrome in current literature worldwide are less than 100 cases. Few previous cases have documented abnormalities of pericardial lesion in patients with ECD, and none of these reports involved the LCH overlap syndrome.
The pathogenesis of ECD and LCH share some similarities. They belong to the “L group” in the histiocytosis classification and have clonal mutations involving genes of the MAPK pathway in over 80% of cases [1]. Studies of genomic analysis have highlighted the structural alterations involving kinases, which activate the MAPK pathway, may be recurrent in L-type histiocytic neoplasms [12]. Mutation of the BRAF V600E gene in MAPK pathway is reported in both ECD and LCH [13]. Since the genetic mutation of BRAF V600E is essential, the primary therapy for these two diseases includes targeted therapy with BRAF inhibition [14,15]. When pericardial lesions are present or central nervous system involvement exists in these two diseases, surgical treatment is also essential.
the best of our knowledge, this is the first reported case of adult ECD and LCH overlap syndrome with pericardial lesion as the first symptom. In this case, the patient presented typical pericardial lesion, diabetes insipidus, and histological evidence showed the infiltration of non-Langerhans cells and Langerhans cells in the tissue. Prior to the diagnosis, the patient underwent regular diagnostic anti-tuberculosis therapy but did not respond well. She was finally diagnosed by immuno-histochemical analysis and genetic testing. The tuberculosis-related testing may be deceptive in the condition, leading to previous underdiagnosis.
Conclusion
In conclusion, these two disorders may not be completely separate entities. This case underlines the necessity to further consider whether there is ECD overlap when diagnosing LCH. Bone scintigraphy can help us distinguish ECD or LDH, and histology can ultimately confirm the diagnosis. Rare disease should be considered when multisystem involvement exists and without tuberculosis or other infection. This case can lead to better understanding of the pathophysiology of both ECD and LCH and help clinicians to confirm diagnosis when encountering rare disorders.
Declarations
Acknowledgements: We thank the participating patient and her families for their contributions.
Funding: This study was supported by the National Multidisciplinary Cooperative Diagnosis and Treatment Capacity Building Project for Major Diseases (Lung Cancer, grant number: z027002).
Conflict of interest: All authors have read and understand the policy of Cancer Science and declare no competing interests.
Ethics statement: Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient to participate in the research.
Consent for publication: Consent for publication in print and electronically has been obtained from the patient for publication of this case report and any accompanying images. All authors have read the manuscript and agree to the publication.
Availability of supporting data: The original contributions presented in the study are included in the article. The data that support the findings of this study are available on request from the corresponding author.
References
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised Classification of Histiocytoses and Neoplasms of the Macrophage-Dendritic Cell Lineages. 2016; 127(22): 2672-81. Epub 20160310. doi: 10.1182/blood-2016-01-690636.
- Wilson MR, Gordon W, Goodlad J, Leach M, Bain BJ. Erdheim-Chester Disease in Bone Marrow. Am J Hematol. 2021; 96(2): 270-1. Epub 20200901. doi: 10.1002/ajh.25948.
- Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for Braf V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data from the Histology-Independent, Phase 2, Open-Label Ve-Basket Study. JAMA Oncol. 2018; 4(3): 384-8. doi: 10.1001/jamaoncol.2017.5029.
- Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med. 2018; 379(9): 856-68. doi: 10.1056/NEJMra1607548.
- Goyal G, Young JR, Koster MJ, Tobin WO, Vassallo R, Ryu JH, et al. The Mayo Clinic Histiocytosis Working Group Consensus Statement for the Diagnosis and Evaluation of Adult Patients with Histiocytic Neoplasms: Erdheim-Chester Disease, Langerhans Cell Histiocytosis, and Rosai-Dorfman Disease. Mayo Clin Proc. 2019; 94(10): 2054-71. Epub 20190828. doi: 10.1016/j.mayocp.2019.02.023.
- McClain KL, Bigenwald C, Collin M, Haroche J, Marsh RA, Merad M, et al. Histiocytic Disorders. Nat Rev Dis Primers. 2021; 7(1): 73. Epub 20211007. doi: 10.1038/s41572-021-00307-9.
- Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester Disease. 2020; 135(16): 1311-8. doi: 10.1182/blood.2019002766.
- Buono A, Bassi I, Santolamazza C, Moreo A, Pedrotti P, Sacco A, et al. Getting to the Heart of the Matter in a Multisystem Disorder: Erdheim-Chester Disease. 2019; 394(10198): 19. doi: 10.1016/s0140-6736(19)31787-8.
- Qanneta R, Raventos-Estelle A. Pulmonary Langerhans-Cell Histiocytosis. N Engl J Med. 2022; 387(26): 2449. Epub 20221224. doi: 10.1056/NEJMicm2203885.
- Krooks J, Minkov M, Weatherall AG. Langerhans Cell Histiocytosis in Children: History, Classification, Pathobiology, Clinical Manifestations, and Prognosis. J Am Acad Dermatol. 2018; 78(6): 1035-44. doi: 10.1016/j.jaad.2017.05.059.
- Courtillot C, Laugier Robiolle S, Cohen Aubart F, Leban M, Renard-Penna R, Drier A, et al. Endocrine Manifestations in a Monocentric Cohort of 64 Patients with Erdheim-Chester Disease. J Clin Endocrinol Metab. 2016; 101(1): 305-13. Epub 20151113. doi: 10.1210/jc.2015-3357.
- Gray JCR, Kim J, Digianvittorio M, Feeley NK, Scheel PJ, Siegelman SS, et al. Braf-Mutated Erdheim-Chester Disease: Profound Response to Vemurafenib Visualized with Serial Multimodality Imaging. J Natl Compr Canc Netw. 2020; 18(6): 650-5. doi: 10.6004/jnccn.2020.7549.
- Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High Prevalence of Braf V600e Mutations in Erdheim-Chester Disease but Not in Other Non-Langerhans Cell Histiocytoses. 2012; 120(13): 2700-3. Epub 20120809. doi: 10.1182/blood-2012-05-430140.
- Abla O, Rollins B, Ladisch S. Langerhans Cell Histiocytosis: Progress and Controversies. Br J Haematol. 2019; 187(5): 559-62. Epub 20190715. doi: 10.1111/bjh.16099.
- Haroche J, Arnaud L, Amoura Z. Erdheim-Chester Disease. Curr Opin Rheumatol. 2012; 24(1): 53-9. doi: 10.1097/BOR.0b013e32834d861d.