
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Understanding bone marrow involvement in Fabry disease: Broader clinical implications explored
Iashvili Levani*; Chavez Yadira; Savariya Badal; Brown Connor; Banerjee Alexander; Rehman Asad; Zhao Bihong; Mai Brenda
The University of Texas Health Science Center at Houston, USA.
*Corresponding Author : Iashvili Levani
The University of Texas Health Science Center at Houston, USA.
Email: levani.iashvili@uth.tmc.edu
Received : Aug 28, 2024
Accepted : Sep 17, 2024
Published : Sep 24, 2024
Archived : www.jcimcr.org
Copyright : © Levani I (2024).
Citation: Marinova L, Georgiev R, Evgeniev N. Clinical observations in three clinical cases with locally advanced chordomas. What is needed for early diagnosis with improved survival?. J Clin Images Med Case Rep. 2020; 1(1): 1005.
Introduction
Epidemiology: Fabry disease is a rare genetic disease that falls under the category of lysosomal storage diseases. The estimated prevalence of Fabry disease in the general population is approximately 1 in 40,000 to 1 in 117,000 individuals. It has been found to be more common in certain ethnic groups, such as populations of Ashkenazi Jewish, African-American, and European descent [1,2].
Pathophysiology: Fabry disease is an X-linked genetic disorder caused by a mutation in the GLA gene, which leads to a deficiency of lysosomal α-galactosidase A (α-GalA). α-GalA is an enzyme responsible for breaking down globotriaosylceramide (Gb3 or GL-3), a glycosphingolipid found in lysosomes. The deficiency of α-GalA leads to the accumulation of glycosphingolipids in the endothelial cells, affecting the kidneys, heart, nervous system, and skin [3].
Clinical presentation
The clinical manifestations of Fabry disease can be classified according to age categories: 1. The Classical presentation of Fabry disease begins in Childhood and Adolescence (≤16 years). In these early years of life, individuals may experience issues with the peripheral nervous system, including paresthesias and neuropathic pain in the extremities. Individuals can have “pain crises” triggered by temperature changes, stress, diseases, or alcohol consumption, indicating small-fiber neuropathy. Autonomic nervous system involvement may result in hypohidrosis, decreased production of saliva and tears, impaired intestinal motility, orthostatic dysregulation, and vertigo. Skin-related manifestations may appear as groups of angiokeratoma, which are often found in areas like the buttocks, around the navel, scrotum, and thighs. Finally, renal and cardiac abnormalities may also become apparent during this stage, including microalbuminuria, proteinuria, and abnormal heart rate variability 2. Early Adulthood (17-30 years): In the initial phase of adulthood, the condition progresses further, and Fabry nephropathy may present with proteinuria and progressive renal insufficiency, which is often accompanied by renal cysts of uncertain origin and hypertension. Another complication is Fabry cardiomyopathy, characterized by left ventricular hypertrophy, conduction disorders such as atrial fibrillation, supraventricular and ventricular tachycardia, valve dysfunction affecting the mitral and aortic valves, angina pectoris, and intramyocardial fibrosis (detectable in cardiac MRI as “late enhancement”). In more rare cases, Fabry may lead to Transient Ischemic Attacks (TIAs), ischemic strokes, rare intracerebral hemorrhages, basilar artery ectasia, white matter lesions seen in cerebral MRI, altered cerebral blood flow, lower extremity lymphedema, depression, and psychoses. 3. Later Adulthood (> 30 years): Renal insufficiency may require interventions such as dialysis or renal transplantation in the later stages of adulthood. Individuals may experience heart failure, malignant arrhythmias, recurrent TIAs and strokes, and the onset of vascular dementia [6]. Regarding gender, Fabry disease is wholly expressed in hemizygous (affected) males, while females are generally thought to have a milder disease course. In, Hemizygous male subjects were, on average, more than 10 years younger than female subjects and generally had more severe pathologic changes on renal biopsy [7,6].
Diagnosis: Diagnosis of Fabry disease is established through clinical presentation, αGalA enzyme activity assays, genetic testing, and, in some cases, kidney biopsy. Specifically, females require genetic testing to diagnose Fabry disease because their presentation of the disease and diagnostic confirmation differ from males. In females, the diagnosis of Fabry disease cannot be confirmed solely by measuring αgalactosidase A (AGAL) activity, as women with Fabry disease often have AGAL activities within the reference range. Instead, molecular genetic testing is necessary to detect disease-causing mutations in the GLA gene for females. This is because females with Fabry disease may have high residual enzymatic activity, making it difficult to diagnose based on AGAL activity levels alone. In addition, we propose using bone marrow biopsy as a supportive method to confirm the disease, especially in cases where hematologic complications are suspected [6].
Treatment: Currently, approved medications for Fabry disease include Enzyme Replacement Therapy (ERT) using Replagal and Fabrazyme, as well as chaperone therapy with migalastat. These therapies reduce the accumulation of Gb3 within cells. ERT involves replacing the missing native AGAL enzyme with recombinant aGAL. Meanwhile, chaperone therapy directly corrects the misfolded native AGAL enzyme via reversible binding to the active site of the misfolded protein, thereby restoring AGAL’s stability. This correction leads to improved transport within cells and increased enzymatic activity within lysosomes. Upcoming therapies include next-generation ERT, gene therapy, and substrate therapy. While ERT has been a vital intervention, developing Antidrug Antibodies (ADAs) in response to ERT medications can hinder their effectiveness. ADAs are antibodies produced by the body’s immune system, which bind to ERT medications and reduce their therapeutic impact. Next-generation ERT therapies are being explored to minimize the development of ADAs, thus enhancing the effectiveness of ERT in managing Fabry disease. Gene therapy delivers DNA containing the AGAL gene into patients’ cells by targeting the defective enzyme at an earlier stage than traditional ERT. Substrate reduction therapies aim to prevent the formation of new Gb3 through de novo synthesis. Supportive treatments aim to manage specific symptoms and complications commonly seen in FD. Pain Management: Neuropathic pain can be treated with pregabalin. Renal Protection: Renin-Angiotensin-System (RAS) blockers, such as ACE inhibitors or ARBs, are used to safeguard the kidneys from renal insufficiency and proteinuria. Gastrointestinal Symptoms: Gastrointestinal issues like cramping, diarrhea, nausea, and bloating can be managed with loperamide, metoclopramide, proton pump inhibitors, or ondansetron. Dietary adjustments and oral AGAL replacement therapy are also options. Cardiac Complications: Cardiac problems, including ventricular tachycardia, heart failure, and coronary stenosis, may require treatment with antiarrhythmics, ICDs, diuretics, ACE inhibitors (or ARBs if ACE inhibitors are intolerable), and statins [2-6].
Case presentation
A 57-year-old male with a past medical history of ESRD on hemodialysis, hypertension, confirmed GLA-X-linked Fabry disease, bilateral ventricular hypertrophy, atrial fibrillation, and aortic and mitral regurgitation presented with anemia, leukopenia, and thrombocytopenia. The patient had a previous history of transient ischemic attack and NSTEMI.
(For reference, cardiac MRI 4/30/23).
Impression
1. Cardiac MRI finding consistent cardiac involvement in Fabry’s disease with evidence of mid to apical cavity obstruction and moderate late gadolinium enhancement burden.
2. Diffusely increased myocardial thickness in the left and right ventricle (maximum thickness of 2.4 cm at mid septum) with reduce global native T1 value (867 msec) that consistent with know Fabry’s disease.
3. Normal left ventricular size and systolic function (LVEF 60%). Cavity obliteration noted at the mid to apical LV segments.
4. Normal left ventricular size and systolic function (RVEF 54%).
5. Patchy late gadolinium enhancement (LGE) involving the septum, lateral and LV apex, likely related to Fabry’s disease. Additionally, there is subendocardial LGE at the apical segment that most likely related to sequale of mid to apical cavity obstruction.Total LGF burden approximately 12% of LV.
6. Mild aortic and mitral regurgitation.
Pathology and ancillary findings
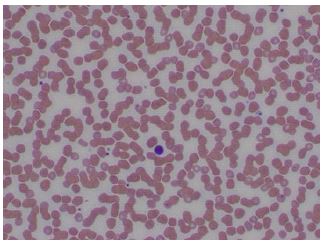
Peripheral blood. The peripheral blood smear showed normocytic normochromic anemia with anisopoikilocytosis and a decreased number of platelets with rare giant forms.
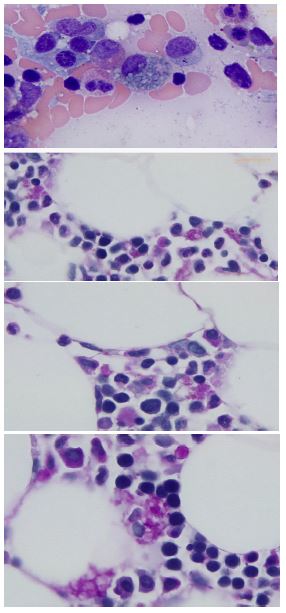
Bone marrow: The bone marrow biopsy demonstrated a mildly hypocellular marrow (25-30%) with rare histiocytes with sea-blue cytoplasmic lipid-like inclusions. Special staining using PAS and PAS with diastase both produced positive results (diastase resistant), which effectively ruled out the possibility of glycogen storage disease. Moreover, these stains accentuated the presence of these inclusions on the bone marrow core biopsy.
Additionally, an elevated number of plasma cells with focal clustering was noted. Subsequent flow cytometry showed no aberrant immunophenotype in the bone marrow sample.
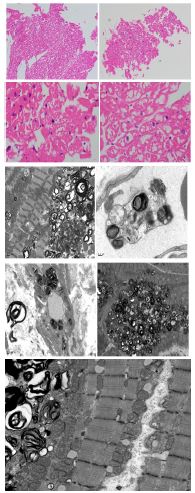
Cardiac tissue: H&E sections in the heart show diffuse, uniform, and severe myocytolysis (vacuolization of the cytoplasm of myocytes) consistent with Fabry’s disease.
The cardiomyocytes exhibit unusual ultrastructural features on electronic microscopy, with numerous myelin figures (laminated bodies) replacing the myocytes. These features correlate with the vacuolization of cytoplasm in light microscopy, which is consistent with Fabry disease. The myelin figures are also seen in interstitial spaces and vascular walls. The unaffected myocytes have central nuclei, and the interstitium contains capillaries and increased amounts of collagen fibrils associated with amorphous material.
Discussion
The biopsy findings in Fabry disease patients corroborate the well-established pathophysiology of the disease. The deposition of Gb3 in various cell types underlies the clinical manifestations observed in affected individuals.
In the context of our case study, a novel avenue was explored by examining a bone marrow biopsy, a departure from the conventional utilization of kidney or cardiac biopsies in the diagnosis of Fabry disease. The presentation of Fabry disease in combination with a plasma cell disorder is rarely reported within the medical literature. The rationale behind performing a bone marrow biopsy was prompted by the patient’s presentation of anemia and thrombocytopenia with giant forms, thus raising suspicion of bone marrow neoplasm. Further examination of bone marrow revealed multiple clusters of plasma cells. However, plasma cells on flow cytometry accounted for only 1.1% of the cells analyzed and showed no abnormal immunophenotype, indicating polyclonally. Thus, with the use of a bone marrow biopsy, we could rule out hematological disease while also being presented with a unique opportunity to gain insights into the bone marrow’s appearance in the context of Fabry disease.
Upon examination, lipid-laden histiocytes were identified in the bone marrow, albeit in limited numbers. To delve deeper into the nature of these cells and their contents, we employed a specialized staining methodology by applying Periodic Acid Schiff (PAS) staining, capable of highlighting both lipids and glycogen, followed by PAS diastase which would break down glycogen, if present. Subsequently, the sustained presence of pink granules following diastase treatment provided tangible evidence that these granules were lipid-based rather than glycogen-based, a finding sparsely documented in the context of Fabry disease.
The heart is one of the major organs affected in FD, leading to cardiac manifestations that can significantly impact patients’ health and prognosis. These cardiac manifestations include ventricular hypertrophy and fibrosis, valve thickening or regurgitation, heart failure, angina, dysrhythmias, cardiac conduction abnormalities, and even sudden death. The clinical manifestation of our patient demonstrated many of these cardiac complications (ventricular hypertrophy and atrial fibrillation atrial and mitral regurgitation with a positive history of TIA and NSTEMI). Of notable significance is the hallmark feature of Fabry disease, the deposition of glycosphingolipids in cardiomyocytes, manifested in the formation of myelin-like structures and concentric inclusions. Electronic Microscopic findings show that these alterations translated to vacuolization within the cytoplasm. The microscopic manifestations of glycosphingolipid deposition in cardiac tissue demonstrate the disease’s multifaceted nature [4,5].
Renal involvement has been documented in case studies and is characterized by podocyte and endothelial cell abnormalities, leading to proteinuria and glomerulosclerosis. The accumulation of Gb3 disrupts cellular function and likely contributes to the progressive renal damage seen in these patients. In advanced cases, when patients have a history of ESRD and hemodialysis, enlargement of the mesangial matrix, global sclerosis of glomeruli, and occasional synechiae may be encountered.
On an Electronic microscope, we can see cytoplasmic distention of podocytes due to lamellated myelin-like inclusions.
Differential diagnosis includes other lipid storage diseases, such as Gaucher’s disease, mucopolysaccharidoses, gangliosidosis, and fucosidosis, and other mimickers of Fabry disease, such as renal diseases and medication-induced phospholipidosis. The association between Myeloid Bodies (MBs), also called “zebra bodies” or “lamellar bodies,” and Fabry disease is well-established. It has long been considered a diagnostic feature of FD; MBs can also be present in patients without FD. Certain medications, such as chloroquine, amiodarone, and aminoglycosides, can cause phospholipidosis with myelin-like figures that mimic Fabry’s disease. In [10], these myelin like figures, characteristic of Fabry disease, are described in patients on Cationic Amphiphilic Drugs (CADs). CADs are drugs that contain a cationic amphiphilic structure capable of inducing phospholipidosis [9,10]. Furthermore, the study reviewed renal biopsies from 32 patients with MBs, of which six had FD. The remaining 26 patients did not have FD but had diverse biopsy findings, including hypertensive arterionephrosclerosis, acute tubular necrosis, and diabetic glomerulosclerosis, among others. In the non-FD group, MBs were primarily observed in podocytes and exhibited a focal pattern [7]. In contrast, the FD group showed more extensive MBs involving various cell types, including podocytes, parietal cells, tubular cells, endothelial cells, and myocytes [2b]. While MBs are commonly associated with FD, they can also be found in other settings, including medications and other inheritable diseases. Therefore, it is important for healthcare professionals to consider clinical history, presentation, and genetics when evaluating patients with symptoms suggestive of Fabry disease.
Conclusion
In conclusion, the meticulous analysis of renal, cardiac, and bone marrow biopsies offers crucial insights into Fabry disease’s intricate morphological changes. These histopathological findings collectively illuminate the systemic impact of Fabry disease and underscore the urgency of early detection. Biopsy-guided insights inform targeted interventions, such as enzyme replacement therapy, to halt irreversible organ damage. Our case study emphasizes the rarity of bone marrow biopsy findings pertaining to Fabry’s disease. The scrutiny of bone marrow contributed to clarifying the diagnostic workup by excluding hematologic disease and unraveling the infrequently observed lipid-laden histiocytes within the scope of Fabry disease. Emphasizing biopsy analyses holds the potential to refine diagnostics and enhance therapeutic strategies for Fabry disease, ultimately improving patient outcomes.
References
- Schiffmann R, Kopp JB. Austin Flint murmur in Fabry disease. 2008; 118(15): 1590-1592.
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. 2001; 345(1): 916.
- Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, et al. Fabry disease, an under-recognized multi systemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. 2003; 138(4): 338-346.
- Umer M, Kalra DK. Cardiac MRI in Fabry disease. Front Cardiovasc Med. 2023; 9: 1075639. doi: 10.3389/fcvm.2022.1075639.
- Azevedo O, Cordeiro F, Gago MF, Miltenberger-Miltenyi G, Ferreira C, et al. Fabry Disease and the Heart: A Comprehensive Review. 2021; 22(9): 4434. doi: 10.3390/ijms22094434.
- Lenders M, Brand E. Fabry Disease: The Current Treatment Landscape. Drugs. 2021; 81(6): 635-645. doi: 10.1007/s40265-021-01486-1.
- Fischer E, Moore M, Lager D. Fabry disease: a morphologic study of 11 cases. 2006; 19: 1295-1301.
- Sangle N. Fabry disease. Pathology Outlines.com website. 2023. https://www.pathologyoutlines.com/topic/kidneyfabry.html.
- Choung HYG, Jean-Gilles J, Goldman B. Myeloid bodies is not an uncommon ultrastructural finding. Ultrastructural Pathology. 2022; 46(1): 130-138. DOI:10.1080/01913123.2021.2022054.
- Shimohata H, Yamashita M, Yamada K, Hirayama K, Kobayashi M. Treatment of Fabry Nephropathy: A Literature Review. 2023; 59(8): 1478.s