
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Series - Open Access, Volume 5
Adrenocortical tumor in children: About three pediatric cases
J Fahri*; M El Baz; J El Houdzi
Department of Pediatrics B, Hôpital Mère-Enfant, CHU Mohammed VI, Marrakech, Morocco.
*Corresponding Author : J Fahri
Department of Pediatrics B, Hôpital Mère-Enfant, CHU Mohammed VI, Marrakech, Morocco.
Email: drjihanefahri@gmail.com
Received : Sep 07, 2024
Accepted : Sep 26, 2024
Published : Oct 03, 2024
Archived : www.jcimcr.org
Copyright : © Fahri J (2024).
Abstract
Adrenocortical tumor is a malignant and rare tumor in children, and are sometimes integrated within the framework of predisposition syndromes such as Li and Fraumeni or Beckwith-Wiedemann syndromes. Their diagnosis is often made on a bundle of clinical, biological, radiological and anatomo-pathological arguments.
Our study was based on three cases of adrenocortical tumor and the object if was to summarize the clinical, biological, radiological features and comprehensive treatment with prognosis of adrenocortical carcinoma in children.
Their treatment essentially involves tumor excision surgery, sometimes supplemented by chemotherapy most often based on platinum salts, etoposide and doxorubicin, combined with mitotane for forms at high risk of recurrence. The role of radiotherapy is discussed, particularly given the frequent involvement of abnormalities in TP53.
The prognosis of these tumors is poor with an overall 5-year survival of less than 30%.
Keywords: Adrenocortical tumor; Pediatric; Chemotherapy; Prognosis; Impact.
Abbreviations: ENSAT: European Network for the Study of Adrenal Tumors.
Citation: Fahri J, El Baz M, El Houdzi J. Adrenocortical tumor in children: About three pediatric cases. J Clin Images Med Case Rep. 2024; 5(10): 3279.
Introduction
Adrenocortical Carcinoma (ACC) is a rare malignancy of unknown pathogenesis and poor prognosis. Foreign data shows that the incidence is about 0.7-2 per million, and patients with adrenocortical tumor account for 0.05-2.00% of patients with malignancy [1]. The independent annual incidence rate of ACC in children is 0.3 per million. Due to the low incidence rate of ACC, there are few reports at this topic what partly explains the knowledge of these tumors.
In adult patients, their histological diagnosis is relatively well codified and the criterias of malignity are well identified. In childhood, these criterias remain discussed, and it is often the evolution of the disease which gives arguments on the characters benin or malignant of these tumors [1,2].
Clinically most children showed clinical signs related to excess steroid hormones or abdominal mass effects while 15% of children are initially diagnosed accidentally.
Aim
We report in this article a retrospective analysis of the cases of children diagnosed with adrenal cortex who were admitted to our service over a period of 7 years “2015-2022” to summarize the clinical, biological, radiological features and comprehensive treatment with prognosis of adrenocortical carcinoma in children.
Case presentations
Chordoma
O.R; 4 years-old; he was admitted for assessment of excessive weight gain during 2 years before.
Clinical exam showed faciotroncular obesity, with BMI at 31,4 and ascites with hyperandrogenism signs (hoarse voice, facial acne, hirsutism and large penis).
Also, he presented hypertension at 18/12 CmHg; facial erythrosis, but no abdominal mass nor lymph nodes.
Biological assessment was carried out which showed hypercortisolemia for 8 hours at 287 ug/l, midnight cortisolemia high at 189 ug/l remained high after braking test, ACTH slowed down to 1.5 ng/l, an increase dehydroepiandrosterone sulfate 774ng/ml, androstenedione increased x5 the normal, increase in dehydroetiandrosterone x2 the normal with an aldosterone/renin ratio of 64.
Abdominal CT scan showed lesion of the right adrenal compartment measuring 19 x 33 x 33 mm without detectable calcification within it with an absolute washout <60% and relative <40% with lower left calicial calculation of 3 x 5 mm density estimated at 285 UH.
Surgical excision of the mass performed and the pathology exam showed a histological appearance of cortico-adrenaloma and a Weiss score noted at 2.
The time between the appearance of clinical signs and the diagnosis was estimated at 2 years.
The treatment of our patient was limited to complete tumor resection surgery without indication to chemotherapy with strict clinical, biological and radiological monitoring every 3 months.
Case 2
M.S; 7 years-old, girl; from a non-consanguineous marriage; youngest of 5 siblings; admitted on 08/06/2015 for abdominal distension evolving for 15 days with fever.
Clinical exam in his admission showed pallor, abdominal mass on the right flank approximately 6 cm in size, with the presence of pubic hair accentuated compared to age “P2” without other signs of precocious puberty and no obesity (weight at 20 kg, BMI at 16,5), blood pressure was normal (100/50 mmhg).
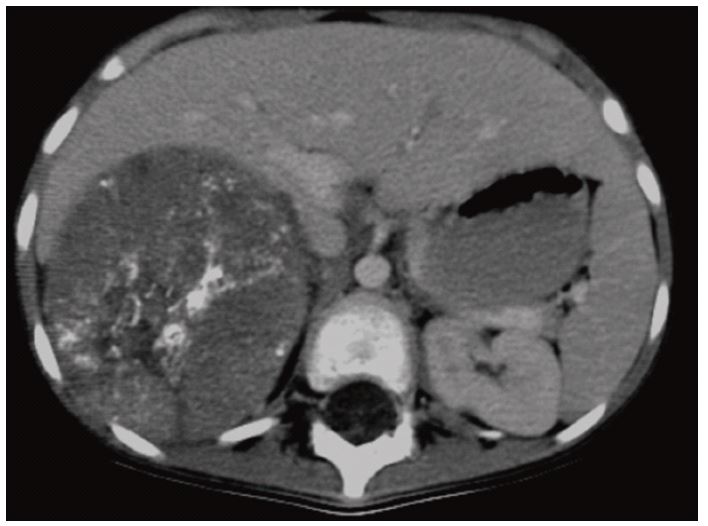
Abdominal CT scan was made and showed a large retroperitoneal right adrenal mass measuring 98/115 cm in favor of a neuroblastoma pushing back the neighboring structures (liver VCI pancreas right kidney) (Figure 1).
A staging assessment was requested with no detectable secondary localization. On the biological assessment, the dosage of catecholamines was normal; adrenaline level<0.02 umol/l (N:<0.10) noradrenaline 0.28 umol/l (N:<0.5) and dopamine 0.72 umol/mmol (N:<0.65).
Urinary free cortisol was 23 ug/24h (N: 10-50 ug/24h) with an aldosterone level increased to 1317 pmol/l (N: 83-405 pmol/l), testosterone at 0.52 ng/ml (N:0.1-0.9 ng/ml) and low 17 beta oestradiol at <9 pg/ml (N: 18-147 pg/ml) delta4andro stenedione increased at 8.9 noml/l (N: 0.3-2.4 nmol/l ) and 17 total cetosteroids increased to 36.36 umol/24h (N: 3.10-10.50 umol/24h).
Patient operated for complete excision of the mass, the pathology exam showed a morphological and immunohistochemical appearance in favor of malignant adrenocortical tumor (Weiss index of 4).
Patient put on chemotherapy CEV/IVE protocol with biological and radiological follow-up.
The CT after Cure 2 was showing total regression of the right adrenal mass with no sign of recurrence or secondary localization. And on the CT scan post IVE 4 appearance of a nodular lesion of the right adrenal lodge 27 x 17 mm infiltrating the upper pole of the right kidney with irregular contours which may be related to a tumor residue or recurrence.
The chemotherapy protocol was maintained and the patient received his 6 courses of chemotherapy.
At the end of the treatment, the control of the abdominal scanner was not carried out the patient died at the age of 8 and a half in an unknown context.
Case 3
Z.E; 5-years-old, boy, youngest of 2 siblings; admitted in March 2014 for dyspnea and oedema at the lower limbs (OMI) evolving for 1 year worsened 1 month ago clinical exam showed dyspnea and hepatomegaly 2 cm of the costal edge with turgidity of the jugular veins and OMI.
Echocardiography was made which objectified an image emerging from the IVC filling the right atrium and engaging the right ventricle compromising the filling of this one in relation to a tumoral extension or thrombus placed on low molecular weight heparin.
An abdominal ultrasound performed which highlights a large mass of the left renal compartment associated with a slight peritoneal effusion with slight right hydronephrosis and nodular lesions of the left liver probably of secondary origin.
Thoraco-abdominal CT showed a retroperitoneal adrenal abdominal mass with multiple adenopathy above mesocolic and left pre-latero-aortic with three small pulmonary nodules evoking a left nephroblastoma.
The patient received 6 courses of preoperative chemotherapy with an almost stable appearance on the CTscan control.
Surgery was done with thrombectomy and a radical nephrectomy. Pathology exam showed malignant cortical adrenaloma with invasion of the renal vein and tumoral vascular emboli.
A biological assessment was revealed to be negative with a free cortisol level at 14 ug/24h with an aldosterone level at 49.6 nmol/l testosterone at 0.13 ng/ml estradiol at 9.15 pg/ml and 17 OH -corticosteroids at 3.30 umol/l
First, patient was not treated by chemotherapy during 6 months but the follow up was not satisfying statement. Patient presented hemoptysis and respiratory discomfort and thoracic and abdominal CT scans showed an increase in the number and size of secondary pulmonary and hepatic nodules with pericarditis and left mediastinal and latero aortic adenopathy with infusion in the left lumbar fossa most likely related to a liquefied hematoma.
Then, chemotherapy protocol CVE / IVE was made. After 3 courses, patient did not present any improvement but progression of lesions and appearance of others sites in bones and lung.
Patient died after several courses of metronomic chemotherapy.
Having received 3 courses of CEV and 2 courses of IVE with CT post IVF 2 liver and lung and bone metastases with latero peritoneal ADP and progressive right adrenal mass. Patient then put on palliative TTT who died in July 2015 at the age of 6 and a half.
Discussion
Adrenocortical Carcinoma (ACC) is a rare malignant tumor, age the onset of the disease is bimodal and occurs mainly in children under five years old and adults between 40 and 50 years old. The disease usually occurs in girls: the male/female ratio is around 1.2. In the present study, two patients were boys (2/3) and one girl. The age of onset was 4.5 to 7.5 years with a median age of onset was 5 years.
Molecular oncogenesis and epigenetic aspects
In the past, advances in identifying the genes involved in ACC came mainly from the study of familial diseases. ACCs were frequently associated with Li-Fraumeni syndrome, due to germline TP53 mutations, and Beckwith-Wiedemann syndrome, due to alterations in the insulin-like growth factor IGF2. At the somatic level, inactivating mutations of TP53 and activating mutations of proto-oncogene β-catenin (CTNNB1) were the most frequent mutations identified in ACC. Recently, through genomic approaches including exon sequencing, not only TP53 and CTNNB1 have been confirmed to be involved in ACC tumorogenesis, but also ZNRF3 (Zinc and ring finger protein 3) was the most frequently altered gene [4]. Additionally, by comparative genomic hybridization (CGH), chromosomal gains at 5, 7, 12, 16, 19, and 20 and losses at 13 and 22 were observed in ACC. Regarding epigenetic changes, a specific CpG island methylation phenotype has been identified in ACC associated with hypermethylation of promoters of specific genes such as H19, PLAGL1, G0S2 and NDRG2. Additionally, some studies have identified significant upregulation of miR-483 associated with downregulation of miR-195 and miR-335 in ACC. Regarding epigenetic changes, a specific CpG island methylation phenotype has been identified in ACC associated with hypermethylation of promoters of specific genes such as H19, PLAGL1, G0S2 and NDRG2. Additionally, some studies have identified significant upregulation of miR-483 associated with downregulation of miR-195 and miR-335 in ACC. Regarding epigenetic changes, a specific CpG island methylation phenotype has been identified in ACC associated with hypermethylation of promoters of specific genes such as H19, PLAGL1, G0S2 and NDRG2. Additionally, some studies have identified significant upregulation of miR-483 associated with downregulation of miR-195 and miR-335 in ACC [2,4,5].
Diagnostic
Diagnosis is based on careful investigations of clinical, laboratory, and imaging features before surgery and pathological examination after tumor removal.
Endocrine assessment
The European Network for the Study of Adrenal Tumors (ENS@T) offers a preoperative hormonal assessment in case of suspected ACC. In particular, the evaluation of basal cortisol, ACTH, dehydroepiandrostenedione sulphate, 17-hydroxyprogesterone, testosterone, androstenedione and estradiol as well as a dexamethasone suppression test and urinary free cortisol are recommended [6]. In recent years there has been growing evidence that some ACCs, previously thought to be non-secreting, may in fact secrete certain urinary steroid metabolites and recently metabolic analysis of urinary steroids has been introduced routinely [6,7].
According to the presence of endocrine function, primary ACC can be divided into two categories: functional ACC mainly produces cortisol, aldosterone and sex hormones. When the tumor volume is small, patients have obvious endocrine disorders and often consult a doctor at the early stage of the tumor, which is the case in 1/3 of our patients. Virilization is the most common symptom in children, accounting for 84.2% of symptoms, manifested by appearance of pubic hair, enlarged clitoris, penis enlargement, facial acne, broken voice, hirsutism, improving muscle development and accelerating growth and development [1,5,7]. Patients with a primary manifestation of Cushing’s syndrome in the infantile age group represent approximately 5%. Symptoms of Cushing’s syndrome include the following: A moon-shaped face, centripetal obesity, and hypertension. Hypertension is the most common symptom [7].
Non-functional ACC does not secrete corticosteroids, so patients generally present with local symptoms and systemic symptoms caused by the tumor, which is the case in 2/3 of our patients. The main symptoms are low back pain, abdominal pain, abdominal mass, loss of appetite, anemia, low fever, bone pain and other non-specific symptoms [5,7]. At the time of diagnosis, the stage is relatively late, with a high degree of malignancy and a poor prognosis, and the natural course of the disease is short. In children, non-functional ACCs account for 10% [8].
Since laboratory tests are non specific, it is necessary to exclude pheochromocytoma, neuroblastoma, and other malignancies when hormone levels are abnormal. Additionally, laboratory examination of endocrine function cannot be used as the primary index to differentiate between benign and malignant tumors because some laboratory indicators change with age [7,9]. This can mask whether the changes are caused by a tumor, making it difficult to make a clinical judgement. Therefore, the laboratory examination is indicative only.
Imaging
Traditional and functional imaging can correctly diagnose an adrenal mass as ACC in most cases. The risk of ACC increases with tumor size, with the index of suspicion increasing for tumors >4 cm (sensitivity, 97%; specificity, 52%) and >6 cm (sensitivity, 91%; specificity, 80%). Unfortunately, masses 1 to 4 cm in diameter are difficult to diagnose [1,4]. Generally, most ACCs are large, heterogeneous with irregular margins. Necrosis, hemorrhage or calcification may be associated.
Currently, no imaging method can characterize a localized adrenal mass as ACC. Regarding traditional imaging, abdominal Computed Tomography (CT) is mandatory in the event of suspected ACC: Many studies have established a threshold of≤10 Hounsfield Unit (HU) in CT without injection for the diagnosis of benign lesion. When the basal density is >10 HU, contrast washout is helpful in distinguishing benign adrenal lesions from ACC. Absolute washout >50% suggests benign adrenal injury. Just as computed tomography is fundamental to defining the stage of the disease, all patients with ACC should perform a chest CT scan to detect lung metastases before surgery [5,7,8].
The state of the art of Magnetic Resonance Imaging (MRI) is less known. In case of suspicion of ACC, when the scanner does not perfectly characterize the adrenal lesion, three major characteristics of the MRI are useful for the diagnosis of ACC [8]: The presence of an isointense to hypointense signal in T1 weighting, a hyper-intense signal in T2-weighted images and a heterogeneous signal drop during chemical shift.
Like chest and abdominal computed tomography, FDG-PET is important for disease staging and prognosis, but its routine use has yet to be validated.
Tumor staging is a widely used tool to assess prognosis in cancer patients. For CCA, the Tumor-Node-Metastasis (TNM) classification proposed by ENS@T (Table 1) is recommended [9,10]. This staging system defines Stages I and II as strictly localized tumors with a size of ≤5 or >5 cm, respectively. Stage III ACCs are characterized by infiltration into surrounding tissues, positive regional lymph nodes, or tumor thrombus in the vena cava and/or renal vein, while stage IV is defined by the presence of distant metastases [10].
Table 1: ENSAT internship at ACC Children.
| Stage | Primary Tumor | Regional Lymph Node | Distant Metastasis |
|---|---|---|---|
| I | T1 | N0 | M0 |
| II | T2 | N0 | M0 |
| III | T1-2 | N1 | M0 |
| T3-4 | N0-1 | M0 | |
| IV | T1-4 | N0-1 | M1 |
Remarks: T1≤5 cm, limited in the adrenal gland; T2>5 cm, limited to the exterior of the adrenal gland; T3 refers to tumors of any size not involved in adjacent organs; T4 refers to tumors of any size involved in adjacent organs; N0 refers to the absence of regional lymph node metastases, N1 refers to the presence of regional lymph node metastases; M0 refers to the absence of distant metastases and M1 to the presence of distant metastases.
Surgery
Surgery is currently the only radical treatment. Drug treatment or local radiotherapy is recommended for patients who have an unresectable tumor, removed risk factors or positive surgical margins. Theoretically, surgery should completely remove all tumors visible to the naked eye and include the removal of surrounding fatty tissue and suspected areas of tumor invasion. For patients with stage IV distant metastases, discuss whether to perform surgery [4,9,11]. However, some researchers believe that resection of the primary tumor may improve prognosis and reduce hormone secretion. Pathology is the gold standard for diagnosis, but no antibodies are specific for the diagnosis of ACC [11].
Pathology
Pathological assessment is the key to the final diagnosis of ACC, but it remains difficult. First, since ACC may be a non-secreting tumor, the adrenocortical origin of the mass must be established. Determination of steroidogenic factor 1 (SF-1) expression has proven to be the most valuable marker. Second, several parameters (macroscopic and microscopic) must be evaluated in order to discriminate benign from malignant tumors.
Macroscopy reveals that ACCs are usually large, heterogeneous, with a surface ranging from brown to orange to yellow depending on the lipid content of their cells. Necrosis is almost always present. Importantly, the presence of tumor invasion at different levels like tumor capsule, extra adrenal soft tissue or direct invasion of lymph ducts, blood vessels are the main hallmarks of ACC [10,12].
Under the microscope, the Weiss score (Table 2) remains the best validated score. It is composed of nine items (three concerning the architecture, three the core and three the presence of any type of invasion) and the presence of an item is worth 1. The sum of the positive items defines the final score. It is established that a Weiss score ≥3 defines ACC, while scores between 0 and 2 define adrenal adenoma, although sometimes a Weiss score of 2 can be suspicious [10,12].
The major problem is the reproducibility of this score and in particular the inter-individual reproducibility. Recently, the practice of the Weiss score by virtual microscopy has been improved by the 12 pathologists of the French adrenal cancer network comete.
Table 2: Weiss score.
| Histological criteria | Degree | Weiss et al (1989) | Items robustes | |
|---|---|---|---|---|
| Atypia nackekite | Moderately severe | 1 | A | |
| Mitoses | >5/50 ИРГ | 1 | A,V | |
| Atypical mitoses | Present | 1 | A | |
| Clear Cells | <25% | 1 | A | |
| Architecture | Diffus is 33% | 1 | ||
| Necrosis | Present | 1 | A,V | |
| Venous invasion | Present | 1 | V | |
| Sinusoidal invasion | Present | 1 | ||
| Capsular invasion | Present | 1 | A | |