
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Clinical Image - Open Access, Volume 5
Bone marrow atypical hemophagocytosis with pancytopenia: Hemophagocytic lymphohistiocytosis or metabolic disorder?
Rihem Mezrigui1*; Saoussen Chouchene1; Nesrine Jammeli2; Jihen Sakouhi1; Bahri Mahjoub2; Mohsen Hassine1
1Hematology Biology, Fattouma Bourguiba University Hospital of Monastir, Monastir 5000, Tunisia.
2Pediatrics Department, Taher Sfar University Hospital of Mahdia, Tunisia.
*Corresponding Author : Rihem Mezrigui
Hematology Biology, Fattouma Bourguiba University Hospital of Monastir, Monastir 5000, Tunisia.
Tel: 216 52 023 615;
Email: rihem_mez@yahoo.fr
Received : Oct 08, 2024
Accepted : Oct 30, 2024
Published : Nov 06, 2024
Archived : www.jcimcr.org
Copyright : © Mezrigui R (2024).
Keywords: Pancytopenia; Inborn errors; Hemophagocytosis; Lysinuric protein intolerance; Bone marrow.
Citation: Mezrigui R, Chouchene S, Jammeli N, Sakouhi J, Mahjoub B, et al. Bone marrow atypical hemophagocytosis with pancytopenia: Hemophagocytic lymphohistiocytosis or metabolic disorder?. J Clin Images Med Case Rep. 2024; 5(11): 3328.
Description
A 10-month-old girl, was admitted to the pediatric department for early-onset pancytopenia. Digestive disorders were noted in her history. Clinically, she was non-dysmorphic and eutrophic, with findings of hepatomegaly and a spleen tip; the rest of the examination was unremarkable.
Laboratory tests revealed pancytopenia with normochromic normocytic anemia (hemoglobin = 10 g/dL), leukoneutropenia (White Blood Cells = 3.1 × 109/L; Neutrophils = 0.8 × 109/L), and thrombocytopenia (Platelets = 111 × 109/L).
Additionally, hepatic cytolysis (Aspartate Aminotransferase = 96 U/L), hemolysis markers (Lactate Dehydrogenase = 1500 U/L, Haptoglobin < 0.1 g/L), elevated ferritinemia (2076 μg/L), and hypernatremia (190 mmol/L) were observed. Immunoglobulin assays, immunological tests, and viral tests were negative. Abdominal ultrasound showed isolated homogeneous hepatosplenomegaly.
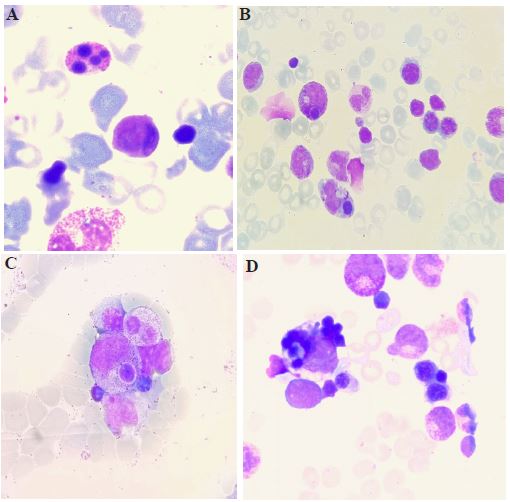
Bone marrow examination revealed a normocellular marrow. There was an increased presence of apoptotic neutrophils (Figure 1A, all images x100, May Grünwald Giemsa stain). Additionally, selective phagocytosis of naked nuclei by immature granular cells was observed (Figure 1B and 1C), an abnormal process where these cells engulfed the nuclei. Numerous hemophagocytic images were present (Figure 1B and 1D), where histiocytes displayed cytoplasm filled with engulfed erythroblasts.
These findings pointed towards a rare metabolic disorder, the lysinuric protein intolerance. This rare condition is characterized by a defective transporter of dibasic amino acids (arginine, ornithine, lysine), leading to their deficient plasma levels and impaired urea cycle function [1]. Diagnosis is confirmed by decreased plasma levels of these amino acids, their increased urinary excretion, and identification of mutations in the SLC7A7 gene [2].
Statements and declarations
The authors declare that our data collection does not require ethics committee approval.
Verbal consent was obtained from the parents before data collection.
Conflict of interest: The authors declare that there is no conflict of interest to disclose.
Funding: The authors declare that the study received no funding.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Olgac A, Yenicesu I, Ozgul RK, et al. Lysinuric protein intolerance: An overlooked diagnosis. Egypt J Med Hum Genet. 2020; 21(42): 1-4. https://doi.org/10.1186/s43042-020-00084-2.
- Noguchi A, Takahashi T. Overview of symptoms and treatment for lysinuric protein intolerance. J Hum Genet. 2019; 64(9): 849-58. https://doi.org/10.1038/s10038-019-0620-6.