
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
A case of Iga MGUS associated leukocytoclastic vasculitis resolved with chemotherapy
Aparajit Ram Venkateswaran; Priya M Reddy*; Peter J Weis; Elina Shustef
Scripps Clinic/Green Rheumatology, San Diego, California, USA.
*Corresponding Author : Priya M Reddy
Scripps Clinic/Green Rheumatology, San Diego, California, USA.
Email: pred81993@gmail.com
Received : Oct 11, 2024
Accepted : Nov 04, 2024
Published : Nov 11, 2024
Archived : www.jcimcr.org
Copyright : © Reddy PM (2024).
Abstract
Monoclonal Gammopathy of Undetermined Significance (MGUS) is a common entity with prevalence increasing with age. While most cases are asymptomatic, a minority of cases may be associated with end organ damage through mechanisms outside of deposition of the monoclonal immunoglobulin or light chain produced by the culprit clonal cell population. Such cases represent the unique clinical entity of monoclonal gammopathy of “clinical” or “cutaneous” significance (MGCS) and have unique therapeutic implications [1-2,10]. Here we present the case of a 35-year-old male with severe cutaneous Leuko cytoclastic Vasculitis (LCV) secondary to Immunoglobulin A (IgA) MGUS with presumptive gastrointestinal vasculitic manifestations that responded to moderate to high dose corticosteroids but failed steroid sparing efforts with dapsone, colchicine, mycophenolate, rituximab and methotrexate. He was treated with cyclophosphamide, bortezomib, and dexamethasone with subsequent normalization of paraprotein production and complete resolution of skin lesions and gastrointestinal symptoms. He remains in remission and off all medical therapy.
Citation: Venkateswaran RA, Reddy PM, Weis JP, Shustef E. A case of Iga MGUS associated leukocytoclastic vasculitis resolved with chemotherapy. J Clin Images Med Case Rep. 2024; 5(11): 3334.
Introduction
MGUS is a common entity with prevalence increasing with age. While most cases are asymptomatic, a minority of cases may be associated with end organ damage through mechanisms outside of deposition of the monoclonal immunoglobulin or light chain produced by the culprit clonal cell population [1]. While dermatologic conditions associated with multiple myeloma are often addressed by the necessary treatment of the underlying malignancy, the decision to treat MGUS and regimen selection remains challenging. Multiple dermatologic associations and recommended treatments have been described with MGCS [1,2], though leukocytoclastic vasculitis associated with IgA gammopathy remains a rare condition with most cases described in association with plasma cell myeloma [3]. Here we describe only the 7th case of LCV associated with IgA monoclonal MGCS [3,4,8,9].
Case presentation
A 34-year-old male with no significant past medical history presented with a four month history of maculopapular, purpuric rash of the lower extremities with progression to his trunk and arms, associated fevers, myalgias, and intermittent abdominal pain. Skin punch biopsy of a right lower extremity lesion revealed perivascular inflammatory infiltrate of neutrophils, lymphocytes and eosinophils associated with prominent fibrin deposition in vessel walls and leukocytoclasis. Immunofluorescence revealed faint granular IgM and C3 in the walls of inflamed superficial dermal vessels, compatible with leukocytoclastic vasculitis. Work up was notable for elevated total protein to 8.6 g/dL with faint bands corresponding to IgA lambda on Serum Protein Electrophoresis (SPEP) and elevated total protein to 17.3 mg/dL without monoclonal proteins on Urine Protein Electrophoresis (UPEP). Serum immunoglobulin level revealed elevated IgA to 897 mg/dL with normal IgG and IgM, consis tent with a diagnosis of presumed IgA vasculitis despite lack of IgA deposition on direct immunofluorescence with initial skin biopsy. Extensive autoimmune and infectious work-up was otherwise negative except for positive ANA (Table 1). He was initially treated with prednisone and topical triamcinolone with some improvement. Dermatology started dapsone followed by colchicine, however the patient continued to report progressive cutaneous lesions. A decision was made to transition to mycophenolate as a steroid-sparing agent, though skin lesions and abdominal pain persisted with inability to taper off prednisone (with significant flare-up of skin lesions with gastrointestinal symptoms below 20 mg per day). Esophagogastroduodenoscopy (EGD) and colonoscopy were performed for evaluation of persistent dyspepsia in the setting of positive fecal occult blood test and ongoing steroid use, revealing mild esophagitis and internal hemorrhoids. Repeat skin biopsy showed continued evidence of leukocytoclastic vasculitis (Figure 1). He received two doses of Rituximab due to recurrent symptoms while continuing mycophenolate. The clinical course was complicated by Varicella Zoster Virus (VZV) meningitis presenting as headache and unilateral hearing loss requiring hospitalization and intravenous antiviral therapy. Computed Tomography (CT) of chest, abdomen and pelvis did not show any significant findings of either infection or neoplasia to explain his refractory disease. After antiviral therapy, his dose of mycophenolate was uptitrated without further benefit. He transitioned to methotrexate without clinical improvement (Figure 2). Immunoglobulin levels were repeated in one year to monitor MGUS, revealing persistently elevated IgA with two monoclonal protein bands on SPEP without accompanying serum light chain elevation (Table 2). UPEP showed a monoclonal free lambda light chain (Bence-Jones proteinuria). Bone marrow biopsy was performed to evaluate for underlying hematologic malignancy, revealing lambda light chain skewed plasmacytosis composing 8-9% of marrow cellularity, consistent with IgA MGCS in the context of skin findings. Flurodeoxyglucose-Positron Emission Tomography (FDG-PET) scan showed intense activity at the gastroesophageal junction corresponding to the area of esophagitis on prior EGD. There was focal intense central prostatic activity which corresponded with a hypervascular nodule on CT with no correlated lesion on prostate magnetic resonance imaging. He was subsequently admitted to the hospital with abdominal pain, vomiting and bloody diarrhea with CT Abdomen and Pelvis revealing enterocolitis which was treated with antibiotics and supportive management. EGD demonstrated esophagitis, which improved with proton pump inhibitor therapy. He was initiated on Cyclophos phamide, Bortezomib and Dexamethasone (CyBorD) for treatment of MGCS per oncology team. He tapered off prednisone and completed four cycles of CyBorD with complete resolution of gastrointestinal symptoms and cutaneous lesions, leaving only mild residual areas of hyperpigmentation. He remains asymptomatic and in remission with normalized IgA and no detectable monoclonal protein more than 8 months after conclusion of chemotherapy.
Table 1: Lab reports.
| LAB | |
|---|---|
| Complete blood cellcount | WBC: 9.7 K/mcL RBC: 4.65 M/mcL Hgb: 13.3 g/dL Hct: 42.6% MCV: 92 fL MCHC: 31.2 g/d LRDW: 13.3% Platelet count: 334 K/mcL Preliminary Abs Neut: 7.32K/mcL Neutrophils: 75.5% IG%: 0.3% Lymphocytes: 15.5% Monocytes: 6.2% Eosinophils: 2.3% Basophils: 0.2% Absolute neutrophils: 7.32% IG#: 0.03 Absolute Lymphocytes: 1.50K/mcL Absolute Monocytes:0.60 K/mcL Absolute Eosinophils:0.22 K/mcL Absolute Basophils: 0.02K/mcL |
| Comprehensive metabolicpanel | Sodium: 138 mmol/L Potassium: 3.9 mmol/L Chloride: 107 mmol/L CO2, Total: 23 mmol/L Glucose:110 mg/dL Urea Nitrogen: 21 mg/dL Creatinine: 1.1 mg/dL Osmolality: 290 Albumin: 4.0 g/dL Protein, total:8.1 g/dL Alkaline Phosphatase: 65 Units/LAST: 15 Units/LALT: 20 Units/L Bilirubin, Total: 0.6mg/dL Anion Gap: 8 mmol/L |
| Urine analysis | Clarity: Clear Specific Gravity: 1.027 pH: 5.5 Protein: Negative Glucose: Negative Ketones: 15 mg/dL Bilirubin: Negative OccultBlood: Negative Leukocyte Esterase: Negative Nitrite: Negative Urobilinogen: 1.0 mg/dL |
| Urine protein and creatinine | Protein:17 mg/dL Creatinine: 118 mg/dL |
| ANA Hep-2Cell, Quant | 1:160, Nucleolar |
| ANCA Vasculitis Panel | Negative |
| C3 | 133 (90-180 mg/dL) |
| C4 | 30.1 (10-40 mg/dL) |
| ESR | 18 (<15 mm/hr) |
| C-reactive protein, high sensitivity | 18.1 (<3.0mg/L) |
| Cryoglobulin | Negative |
| Immunoglobulins IgG, IgA, IgM | Immunoglobulin G: 1,050(700-1,600 mg/dL) Immunoglobulin M: 110 (40-230 mg/dL) Immunoglobulin A: 897 (70-400 mg/dL) |
| SPEP | Protein,total: 8.6 (6.3-8.2 g/dL) Albumin: 3.3-4.4 g/dL Alpha 1: 0.3 (0.1-0.3 g/dL) Alpha 2: 1.0 (0.4-1.0 g/dL) Beta:1.5 (0.8-1.3 g/dL) Gamma Globulin: 1.3 (0.8-1.7 g/dL) Interpretation: There is a band of restricted migration that partially overlies transferrin in the beta region and involves the beta-gamma interface. Immunofixation shows faint bands corresponding to IgA lambda with no suppression of polyclonal immunoglobulins. This findingraises the possibility of a monoclonal gammopathy ortransient immune reaction. |
| Electrophoresis, urine | Protein,Total (UR):17.3 (<11.1 mg/dL) Albumin (UR)L 4.1Gamma Globulin: 13.2 Protein Electrophoresis Interpretation: No monoclonal proteins are identified |
| Hepatitis | Hepatitis B coreantibody, total: non reactive Hepatitis B Surface Antibody: immune to HBV infection Hepatitis B Surface Antigen: non reactive Hepatitis C Antibody: non reactive |
| Quantiferon TB | Negative |
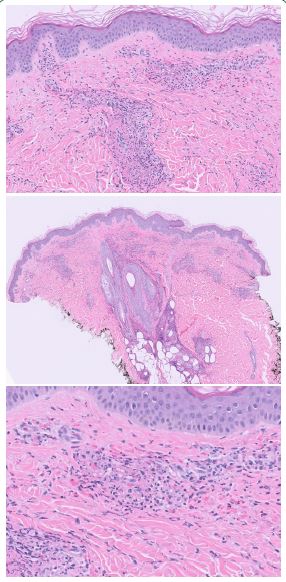
5x: Superficial to mid dermal perivascular inflammatory infiltrate.
20x: Fibrinoid necrosis of vessel walls within the superficial dermis with surrounding extravasated erythrocytes.
40x: Destruction of vessel walls with surrounding neutrophils and associated leukocytoclasis.
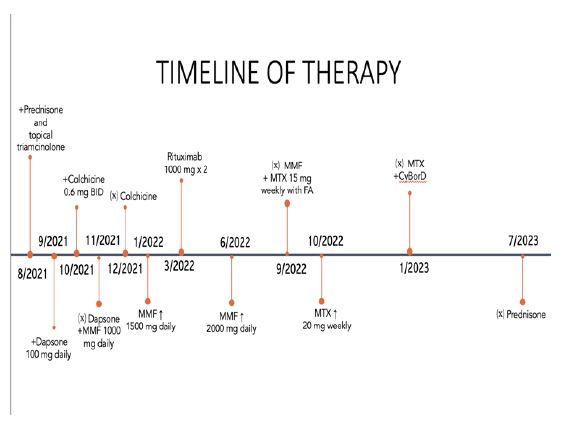
8/2021: Initiation of prednisone and topical triamcinolone.
9/2021-11/2021: Dapsone 100 mg daily. 10/2021- 12/2021: Colchicine.
11/2021: Mycophenolate mofetil 1000 mg daily, increased to 1500 mg daily 1/2022-6/2022, increased to 2000 mg. daily from 6/2022- 9/2022.
3/01/2022, 3/15/2022: Rituximab 1000 mg IV. 9/2022- 1/2023: Methotrexate.
Table 2: Serum protein electrophoresis monitoring.
| 12/2021 | Protein, total: 7.2 Albumin: 3.8 Alpha 1 : 0.2 Alpha 2: 1.0 Beta: 1.3 Gamma Globulin: 0.9 There is a faint protein band present in the beta region with immunofixation electrophoresis that is composed of IgA lambda immunoglobulin. COMMENT: The findings remain non-diagnostic. |
| 4/2022 | Protein, total: 7.4 Albumin: 4.1 Alpha 1: 0.2 Alpha 2: 1.0 Beta: 1.2 Gamma globulin: 0.9 Monoclonal protein: 0.2 (elevated) There are two monoclonal protein bands present. One is in the gamma region and immunofixation electrophoresis shows an IgA band without an accompanying light chain, and the second band overlies transferrin in the beta region showing a faint IgA lambda band similar to previous studies dated 12/29/21. There is a mild suppression of polyclonal immunoglobulins. |
| 9/2022 | Protein, total: 7.6 Albumin: 3.7 Alpha 1: 0.3 Alpha 2: 1.1 Beta: 1.3 Gamma globulin: 1.2 Monoclonal protein: 0.2 There are two monoclonal protein bands present. One is in the gamma region and immunofixation electrophoresis shows an IgA band without an accompanying light chain. The second band overlies transferrin in the beta region showing a faint IgA lambda band similar to previous studies. There is mild suppression of polyclonal immunoglobulins. |
| 1/2023 | Protein, total: 6.8 Albumin: 3.5 Alpha 1: 0.2 Alpha 2: 1.0 Beta: 1.2 Gamma globulin: 0.9 There are two monoclonal protein bands present. One is in the gamma region and immunofixation electrophoresis shows an IgA band without an accompanying light chain. the second band overlies transferrin in the beta region showing a faint IgA lambda band similar to previous studies. There is mild suppression of polyclonal immunoglobulins. |
| 3/2023 | Protein, total: 6.0 Albumin: 3.5 Alpha 1: 0.2 Alpha 2: 0.9 Beta: 0.9 Gamma globulin: 0.4 Hypogammaglobulinemia. No definitive serum monoclonal proteins are identified with immunofixation. |
Discussion
Leukocytoclastic vasculitis is a small vessel, immune-mediated cutaneous vasculitis of the dermal capillaries and venules [5]. It is histopathologically characterized by neutrophilic infiltration of the vessel wall with leukocytoclasia and fibrinoid necrosis [6]. Classically, patients present with non-blanching, palpable purpura and petechiae in dependent areas, most commonly the bilateral lower extremities. It can be idiopathic in nearly half the cases or associated with exposure to certain drugs, infections, malignancy, systemic autoimmune disease and vaccinations. Paraneoplastic leukocytoclastic vasculitis has been observed in leukemia, lymphoma and solid tumors such as squamous cell carcinoma of lung and intestinal adenocarcinoma [7]. It typically confers a worse prognosis and may manifest weeks, months, or years before malignancy is diagnosed [7]. History and physical exam with the appropriate laboratory studies to evaluate for infection, malignancy and autoimmune disease is essential for the work up of leukocytoclastic vasculitis. Diagnosis is made via skin biopsy and should include both the superficial dermis and hypodermis with ideal timing recommended within 48 hours of lesion onset. The primary histologic characteristics seen on biopsy include destruction of dermal small blood vessels by neutrophilic inflammatory infiltrates with leukocytoclasia, fibrinoid necrosis and extravasation of red blood cells [6]. These samples should also undergo testing for direct immunofluorescence as was done in our patient. In this case, serologies supported the diagnosis of presumptive IgA vasculitis though IgA deposition was not observed on direct immunofluorescence in two skin biopsies. In cases where a secondary cause is identified, management strategy should focus on eliminating the trigger or underlying disease process. In idiopathic LCV with skin-limited disease with mild symptoms, conservative treatment is recommended with rest, elevation and compression. When episodes recur or if symptoms are severe, systemic corticosteroids are recommended. There is no proposed standard initial dose for steroids, though higher doses are often used in more severe disease (0.5-1 mg/kg/day prednisone equivalents). The duration of treatment is between 3-6 weeks on average. In patients with persistent disease activity or relapse following steroid discontinuation, colchicine or dapsone is administered after steroids. Systemic immunosuppressive therapy is employed in cases refractory to colchicine and dapsone including methotrexate, mycophenolate mofetil, azathioprine, cyclosporine and rituximab [6].
LCV associated with IgA gammopathy remains a rare condition with most cases described in association with IgA myeloma in the literature [3], though our case is only the seventh associated with IgA MGUS/MGCS [3,4,8,9]. MGCS is typically treated when organ damage related to the secreting clone is detected via laboratory or clinical presentation [3]. Decision to treat can be challenging in cases without a direct causal link between the clone and clinical findings, as in our case, in which IgA was not present on direct immunofluorescence despite presence of concurrent monoclonal gammopathy. Paule, et al successfully treated a similar patient with IgA associated LCV and 10% plasma cell infiltrate with lenalidomide, bortezomib, and dexamethasone as the higher level of plasma cell infiltrate was concerning for smoldering myeloma. We selected CyBorD due to precedent in other MGCS and to avoid lenalidomide given no evidence of smoldering myeloma and potential increased long term risk for secondary malignancies in this young patient [1]. Our case highlights the importance of recognition of IgA MGCS as a unique clinical entity and the efficacy and tolerability of CyBorD for its treatment, particularly in young patients or those with additional renal abnormalities.
References
- Fermand JP, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, et al. Monoclonal gammopathy of clinical significance: A novel concept with therapeutic implications. Blood, the Journal of the American Society of Hematology. 2018; 132(14): 1478-1485.
- Claveau JS, Wetter DA, Kumar S. Cutaneous manifestations of monoclonal gammopathy. Blood cancer journal. 2022; 12(4): 58.
- Umemura H, Yamasaki O, Iwatsuki K. Leukocytoclastic vasculitis associated with immunoglobulin a lambda monoclonal gammopathy of undetermined significance: A case report and review of previously reported cases. The Journal of Dermatology. 2018; 45(8): 1009-1012.
- Paule R, Vignon M, Régent A, London J, Cohen P, et al. French Vasculitis Study Group. IgA monoclonal gammopathy associated with refractory IgA vasculitis successfully treated with clone-targeted therapy. Autoimmunity Reviews. 2020; 19(9): 102611.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic Vasculitis. In: StatPearls. Treasure Island (FL): StatPearls Publishing. 2023.
- Fraticelli P, Benfaremo D, Gabrielli A. Diagnosis and management of leukocytoclastic vasculitis. Intern Emerg Med. 2021; 16: 831-841. https://doi.org/10.1007/s11739-021-02688-x.
- Rabin SS, Dudelzak J, Lee JR, Sheehan DJ. Leukocytoclastic vasculitis: a marker of underlying malignancy. Consultant. 2006; 46(10): 1196-1198.
- Rousset L, Cordoliani F, Battistella M, et al. Vasculitis and IgA monoclonal gammopathy of cutaneous significance. J Eur Acad Dermatol Venereol. 2018; 32: e175-e176.
- Drerup Christian, et al. Evidence for immunoglobulin-mediated vasculitis caused by monoclonal gammopathy in monoclonal gammopathy of unclear significance prompting oncologic treatment. JAAD Case Reports. 2019; 5(3): 288-291.
- Lipsker D. Monoclonal gammopathy of cutaneous significance: Review of a relevant concept. Journal of the European Academy of Dermatology and Venereology. 2017; 31(1): 45-52.
- Oganesyan A, Gregory A, Malard F, Ghahramanyan N, Mohty M, et al. Monoclonal gammopathies of clinical significance (MGCS): In pursuit of optimal treatment. Front Immunol. 2022; 13: 1045002. doi: 10.3389/fimmu.2022.1045002.