
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
A case of sacrococcygeal teratoma of a three-year children
Samuel Mbozo’o Mvondo1*; Yannick Richard Onana1; Aminou Mohamadou1; Jérémie Mbo Amvene1; Boniface Moifo2; Odile Fernande Zeh2; David Ngaroua1
1Faculty of Medicine and Biomedical Sciences of Garoua, University of Garoua, Garoua, Cameroon.
2Faculty of Medicine and Biomedical Sciences, University of Yaoundé 1, Yaoundé, Cameroon.
*Corresponding Author : Samuel Mbozo’o Mvondo
Faculty of Medicine and Biomedical Sciences of Garoua, University of Garoua, Garoua, Cameroon.
Email: smbozoo@hotmail.com
Received : Oct 09, 2024
Accepted : Nov 04, 2024
Published : Nov 11, 2024
Archived : www.jcimcr.org
Copyright : © Mbozo’o Mvondo S (2024).
Abstract
We report a rare case of a 3-year-old boy who presented with multiple pelvi-iliac masses and a mixed tissular and cystic retrosacrococcygean mass. The diagnosis was confirmed by Computed Tomography (CT) scan and Magnetic Resonance Imaging (MRI). The patient underwent surgical resection of the masses and histopathological examination revealed a diagnosis of Sacrococcygeal Teratoma (SCT). The patient had an uneventful postoperative course and was discharged after 10 days. We discuss the clinical features, radiological findings, and management of this uncommon tumor.
Citation: Mvondo SM, Onana YR, Mohamadou A, Amvene JM, Moifo B, et al. A case of sacrococcygeal teratoma of a three-year children. J Clin Images Med Case Rep. 2024; 5(11): 3335.
Introduction
SCT is a germ cell tumor that arises from the presacral region and can extend into the pelvis, abdomen, or retroperitoneum. It is the most common congenital neoplasm in neonates and infants, with an incidence of 1 in 35,000 to 40,000 live births [1]. SCT has a female predominance of 3:1 to 4:1 [2]. SCT can be classified into four types according to the Altman classification: type I (external), type II (predominantly external with minimal presacral component), type III (predominantly internal with significant presacral component), and type IV (completely internal) [3]. The prognosis of SCT depends on several factors, such as tumor size, histological type, presence of malignancy, and completeness of surgical resection [4]. We present a rare case of a 3-year-old boy with multiple pelvi-iliac masses and a mixed tissular and cystic retrosacrococcygean mass that was diagnosed as SCT.
Case presentation
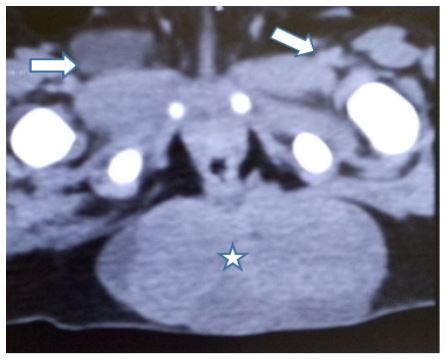
A 3-year-old boy was referred to our hospital with a history of progressive abdominal distension and constipation for 6 months. He had no history of fever, weight loss, or urinary symptoms. His antenatal and perinatal history was unremarkable. His physical examination revealed a palpable mass in the lower abdomen extending to the pelvis. His vital signs were stable and his laboratory tests were normal. He underwent a CT scan that showed multiple well-defined heterogeneous masses in both iliac fossae and a large mixed tissular and cystic mass in the retrosacrococcygean region measuring 4 cm on its long axis (Figure 1). The masses displaced the bladder anteriorly and compressed the rectum posteriorly. There was no evidence of calcification, necrosis, or hemorrhage within the masses. The CT scan also showed bilateral inguinal lymphadenopathy. The differential diagnosis included lymphoma, neuroblastoma, rhabdomyosarcoma, and SCT.
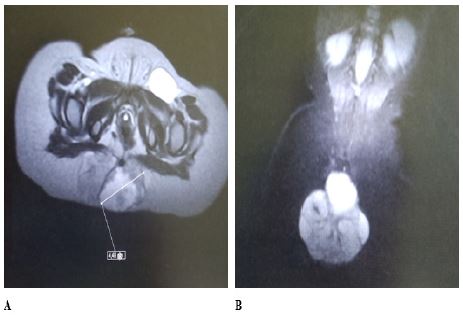
The patient underwent an MRI that confirmed the findings of the CT scan and showed that the masses had high signal intensity on T2-weighted images and low signal intensity on fat-suppressed images.
The patient underwent surgical exploration through a midline laparotomy incision. The intraoperative findings revealed multiple encapsulated masses in both iliac fossae that were easily separated from the surrounding structures. The masses were excised completely along with the inguinal lymph nodes. The retrosacrococcygean mass was also excised completely with a margin of normal tissue. The mass was attached to the coccyx, which was also removed. The total weight of the resected masses was 1 kg. The postoperative period was uneventful and the patient was discharged after 10 days.
The histopathological examination of the masses showed features of SCT with immature and mature elements from all three germ layers, such as cartilage, bone, neural tissue, respiratory epithelium, and glandular structures. The tumor had a benign appearance with no evidence of malignancy or Teratoma with Malignant Transformation (TMT). The inguinal lymph nodes were negative for tumor involvement.
The patient was followed up regularly with clinical examination and imaging studies. He had no evidence of recurrence or metastasis at 2 years of follow-up.
Discussion
SCT is a rare tumor in children older than 2 years, accounting for less than 10% of all cases [5]. The majority of SCTs are diagnosed prenatally or in the neonatal period. The delayed presentation of SCT may be due to the slow growth of the tumor, the lack of specific symptoms, or the misdiagnosis as other abdominal or pelvic masses [6]. The diagnosis of SCT is based on the clinical presentation, the radiological findings, and the histopathological confirmation. The imaging modalities that are useful for the diagnosis and staging of SCT include ultrasound, CT scan, and MRI. Ultrasound can detect the presence, size, location, and composition of the tumor. CT scan can provide more detailed information about the extent of the tumor, its relation to adjacent structures, and the presence of calcification or metastasis. MRI can better delineate the soft tissue components of the tumor and its vascular supply [7].
The treatment of choice for SCT is complete surgical resection with preservation of normal structures and function. The surgical approach depends on the size, location, and type of the tumor. For type I and II tumors, a posterior sagittal approach may be sufficient. For type III and IV tumors, a combined anterior and posterior approach may be required [8]. The coccyx should be removed along with the tumor to prevent recurrence [9]. Adjuvant chemotherapy may be indicated for malignant or recurrent tumors [10].
The prognosis of SCT depends on several factors, such as tumor size, histological type, presence of malignancy, and completeness of surgical resection. The overall survival rate for benign SCT is 90-95%, while for malignant SCT it is 40-60% [11]. The most common complications of SCT include recurrence, infection, hemorrhage, urinary or bowel dysfunction, and sacral deformity [12].
Conclusion
We report a rare case of a 3-year-old boy with multiple pelvi-iliac masses and a mixed tissular and cystic retro sacrococcygean mass that was diagnosed as SCT. The patient underwent successful surgical resection of the masses and had a good outcome. SCT should be considered in the differential diagnosis of abdominal or pelvic masses in children.
References
- Altman RP, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974; 9: 389-98.
- Rescorla FJ. Pediatric germ cell tumors. Semin Pediatr Surg. 1999; 8: 160-71.
- Altman RP. Sacrococcygeal teratomas: diagnosis and management. Curr Probl Surg. 1980; 17: 345-400.
- Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993; 21: 172-81.
- Grosfeld JL, Billmire DF. Teratomas in childhood: analysis of 142 cases. J Pediatr Surg. 1985; 20: 548-51.
- Bajpai M, Pal K, Gupta DK. Delayed presentation of sacrococcygeal teratoma: An experience from a tertiary care center in India. J Indian Assoc Pediatr Surg. 2010; 15: 121-4.
- Daneman A, Lobo E, Alton DJ, Shuckett B. Sacrococcygeal teratomas: Prenatal diagnosis by sonography and magnetic resonance imaging. Pediatr Radiol. 1998; 28: 101-7.
- Pena A. Surgical management of anorectal malformations: a unified concept. Pediatr Surg Int. 1989; 4: 222-31.
- Heerema-McKenney A, Harrison MR, Bratton B, et al. Congenital teratoma: A clinicopathologic study of 22 fetal and neonatal tumors. Am J Surg Pathol. 2005; 29: 29-38.
- Rescorla FJ, Sawin RS, Coran AG, et al. Long-term outcome for infants and children with sacrococcygeal teratoma: A report from the Childrens Cancer Group. J Pediatr Surg. 1998; 33: 171-6.
- Isaacs H Jr. Perinatal (fetal and neonatal) germ cell tumors. J Pediatr Surg. 2004; 39: 1003-13.
- Gobel U, Schneider DT, Calaminus G, et al. Germ-cell tumors in childhood and adolescence. GPOH MAKEI and the MAHO study groups. Ann Oncol. 2000; 11: 263-71.