
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 5
Antenatal diagnosis of recurrent pontocerebellar hypoplasia type 12 associated with common Indian mutation in COASY gene: Importance of testing the index child
Vandana Bansal1*; Nishigandha Mali1; Parag Tamhankar1; Palak Sharma2*
1Department of Obstetrics, Gynecology, and Fetal Medicine, Nowrosjee Wadia Maternity Hospital, and Seth G.S. Medical College, Parel, Mumbai, Maharashtra, India.
2D.Y. Patil University- School of Medicine, Navi Mumbai, India.
*Corresponding Author : Vandana Bansal
Professor, Department of Obstetrics, Gynecology,
and Fetal Medicine, Nowrosjee Wadia Maternity
Hospital, and Seth G.S. Medical College, Parel,
Mumbai, Maharashtra, India.
Email: drvandana_bansal@gmail.com
Received : Nov 24, 2024
Accepted : Dec 12, 2024
Published : Dec 19, 2024
Archived : www.jcimcr.org
Copyright : © Bansal V (2024).
Abstract
Background: Congenital anomalies affect 2-3% of all live born babies worldwide. Pontocerebellar hypoplasia is a group of rare congenital central nervous system malformations with an associated underlying genetic etiology.
Case summary: A fourth gravida had a history of three neonatal deaths; her last two babies had pontocerebellar hypoplasia. Whole exome sequencing of the last affected sibling identified a biallelic COASY (coenzyme A synthase) gene mutation, for which both parents were found heterozygous. This facilitated early invasive testing and diagnosis in the subsequent pregnancy.
Clinical significance: Ten percent of all congenital anomalies have a chromosomal etiology and about 8 percent are monogenic disorders. Pontocerebellar hypoplasia (PCH) is a group of malformations characterized by progressive microcephaly, spasticity, limb contractures, neuro-developmental and psychomotor delay and early neonatal death. Identification of several pathogenic genes and subtle phenotypic variations has helped classify PCH subtypes. Identification of the implicated gene by testing the index child helps in prognostic counseling of possible outcomes and guide management of further pregnancies.
Invasive testing at 11-13 weeks gestation helps look for the pathogenic mutations and can be vital for decision making even before the fetus presents phenotypic features on ultrasound. Here we describe a case of COASY mutation, now considered a founder mutation in the South Asian population for pontocerebellar hypoplasia type 12.
Keywords: Pontocerebellar hypoplasia; Congenital anomalies; Prenatal diagnosis; COASY gene.
Citation: Bansal V, Mali N, Tamhankar P, Sharma P. Antenatal diagnosis of recurrent pontocerebellar hypoplasia type 12 associated with common Indian mutation in COASY gene: Importance of testing the index child. J Clin Images Med Case Rep. 2024; 5(12): 3394.
Background
Worldwide, congenital anomalies affect 2-3% live born and 20% stillborn infants [1]. While a quarter of global neonatal deaths occur in India, congenital anomalies constitute the fifth largest cause of neonatal mortality [2,3]. The prevalence of congenital anomalies in the country is 6-7% which translates to about 1.7 million cases annually [4]. Despite these numbers, socioeconomic constraints often preclude a detailed search for a genetic cause even in cases with multiple affected members in the same family or recurring anomalies in subsequent pregnancies. We present a case of recurrent pontocerebellar hypoplasia in siblings, where genomic analysis by exome sequencing in the index child identified the pathogenic mutation. This aided early prenatal testing in the next pregnancy.
Case presentation
A 27 year old woman in a third-degree consanguineous marriage conceived for the fourth time. She lost three babies in the neonatal period due to central nervous system malformations.
Antenatal care was suboptimal in her first pregnancy in 2014. A term male child delivered by caesarean section for obstetric indications died on third day of life. No definitive cause was discovered.
Late third trimester ultrasound in her second pregnancy (2019) diagnosed fetal cerebellar hypoplasia. This fetus was evaluated by amniocentesis and karyotyping yielded normal results, but financial constraints prevented further genomic analysis and the DNA was saved for future testing. The term baby died in early neonatal period due to respiratory complications. The couple was advised regarding strong possibility of a genetic etiology but they chose to defer further investigations until next pregnancy.
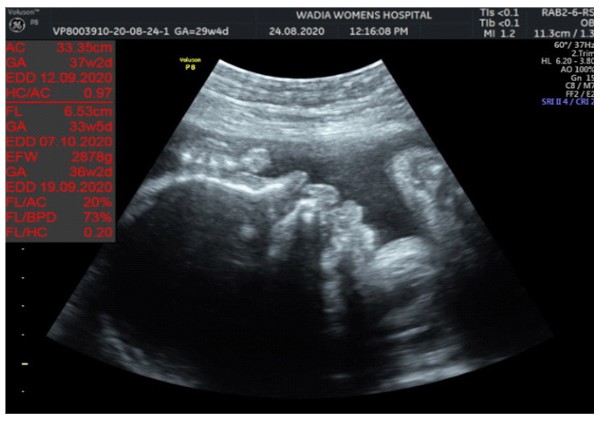
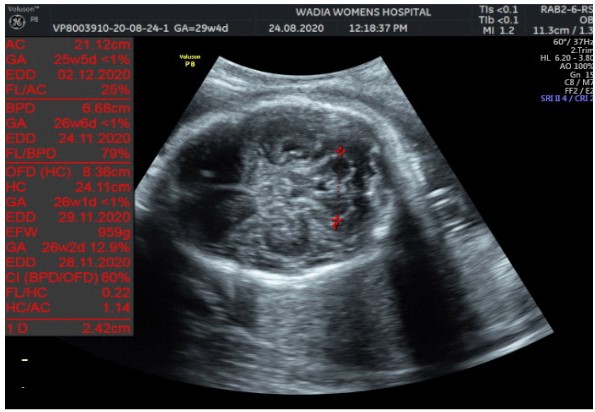
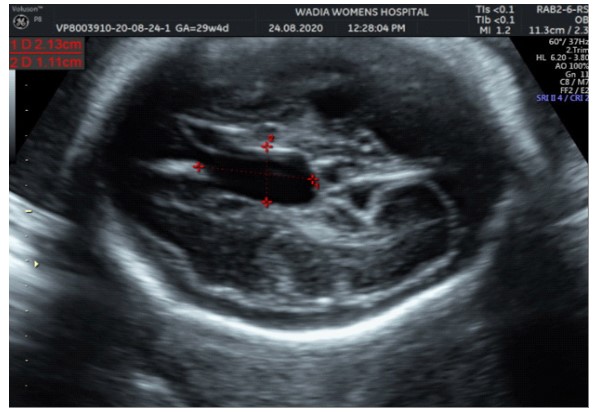
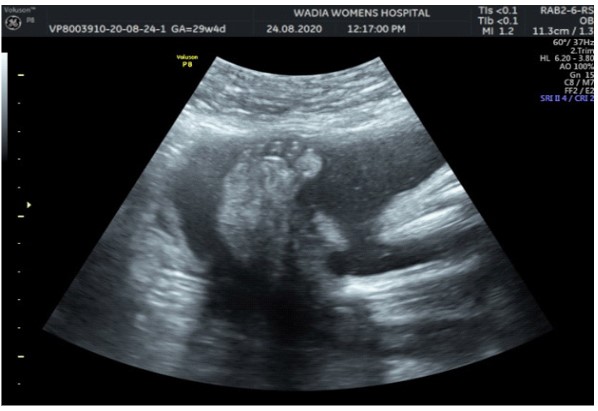
In the third conception during the COVID-19 pandemic, the couple presented to us in the third trimester. Anomaly screening ultrasound at 18 weeks done elsewhere was unremarkable, but subsequent scan done at our institute at 29 weeks was suggestive of fetal biometry corresponding to 26 weeks, head circumference below 1st centile, composite growth along 3rd centile. The fetus had microcephaly and sloping forehead; head circumference measured 241 mm (less than 1st centile for gestational age) (Figure 1). Transcerebellar diameter measured 23.6 mm (<1st centile) with vermis spared suggestive of cerebellar hypoplasia (Figure 2). The Cavum Septum Pellucidum (CSP) was enlarged measuring 22 mm x 10 mm (Figure 3). Additional ultrasound findings were corpus callosum agenesis, kinked thalami, widened subdural spaces and right club foot (Figure 4). The ultrasound differentials were pontocerebellar hypoplasia or Walker Warburg syndrome.
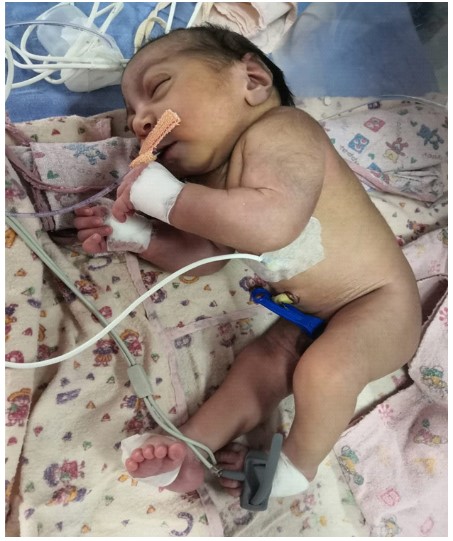
Due to advanced gestation the couple opted to test this fetus postnatally. A term male child weighing 1.98 kg poor Apgar scores (4/10 and 5/10 at 1 and 5 minutes). The neonate had microcephaly, symmetric growth restriction, abnormal flexion of all limbs (arthrogryposis), right congenital talipes equinovarus. This baby required mechanical ventilation since birth and succumbed on day 19 of life (Figure 5).

Investigations: Whole exome sequencing in the third baby postnatally identified a homozygous known variant chr17:40717674C>G (GRCh37 or hg19 format) or c.1486-3C>G, depth 207x that affects the position 3 nucleotides upstream of the acceptor splice site of the exon 9 (transcript number NM_025233.7). This variant has been previously reported in patients affected with pontocerebellar hypoplasia type 12, microcephaly and arthrogryposis. This variant has a minor allele frequency of 0.04%, 0.009% and 0.08% in the 1000 genomes, gnom AD and internal databases of Medgenome Labs Pvt Ltd (Bengaluru, India) respectively. The insilico prediction of the variant is damaging by MutationTaster2 software. The reference base is conserved across species. The variant is also reported as likely pathogenic in Clinvar database (https://www. ncbi.nlm.nih.gov/clinvar/variation/599341/). As per American College of Medical Genetics (ACMG) guidelines of classification of variants, the following criteria are met: PM3 criteria for recessive disorders, detected in trans with a pathogenic variant, PM2 criteria absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium, PP3 criteria Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.), PP5 criteria reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation.
Sanger sequencing of the parents’ blood samples revealed they were heterozygous for the same mutation. The couple was counseled regarding a 25% recurrence risk (autosomal recessive) in all future pregnancies. The options of early invasive testing in each pregnancy or using donor gametes were discussed.
The patient conceived her fourth pregnancy in 2021. The first trimester ultrasound for aneuploidy screening was normal. Chorionic villus sampling was performed at 12 weeks and fetus tested heterozygous for COASY mutation and likely to be unaffected (Figure 6).
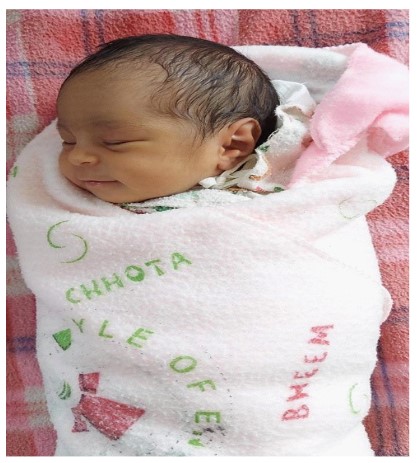
The couple was counseled regarding the carrier status of this fetus and opted to continue pregnancy. Detailed anatomical ultrasound at 18 and 28 weeks of gestation were within normal limits. She delivered a healthy male child at term weighing 3 kg. This baby’s gross appearance, tone, respiration, feeding and postnatal MR imaging were normal (Figure 7).
Discussion
Pontocerebellar Hypoplasia (PCH) is a group of central nervous system malformations with prenatal onset. It is characterized by progressive microcephaly with marked hypoplasia of cerebellum leading to seizures, muscular tone abnormalities, spasticity, extrapyramidal symptoms, limb contractures, neurodevelopmental and psychomotor delay. Death in early neonatal period is common and often due to respiratory complications. The pathology of pontocerebellar hypoplasia was first reported in 1961 in an autopsy specimen of a boy suspected to have Werdnig Hoffman disease [5]. Further reports continued to describe cases with combined features of Spinal Muscular Atrophy (SMA) and olivopontocerebellar hypoplasia [6,7]. It was only in 2003 that Pontocerebellar Hypoplasia (PCH) was considered as a separate entity when gene sequencing did not find abnormalities in the SMN gene [8]. Since then, several subtypes of PCH have been described and variants in 27 genes are implicated till date [9]. The COASY gene is located on chromosome 17q21.2. Its codes for Coenzyme A (CoA) synthase, a bifunctional enzyme essential for the last two steps of Coenzyme A synthesis. Coenzyme A is an essential cofactor as well as substrate for several key metabolic processes involving pyruvate dehydrogenase, 2-ketoglutarate dehydrogenase, acyl-CoA synthase and is required for fatty acid oxidation, citric acid cycle and amino-acid catabolism. CoA has also now been implicated in cell signaling and DNA integrity [10-13]. Animal models have shown that a disruption in CoA synthesis pathways affects lipid biosynthesis, DNA integrity and cellular survival. This in turns manifests as neurodegeneration, impaired locomotor function and reduced lifespan. The COASY gene has been implicated in two neurological disorders: neurodegeneration with brain iron accumulation (NBIA) and pontocerebellar hypoplasia [14,15]. The first report of COASY gene variants in PCH was by van Djik et al in 2018 who described four individuals from two families who were either homozygous (c.1486-3 C>G) or compound heterozygous (c.[1549_1550delAG]; [1486-3 C>G]) for variants in the COASY gene [16]. An Indian series describes nine individuals from five families with PCH 12 having the biallelic mutation COASY: c.1486-3C>G variant which is the same variant as identified by our case [17]. All the individuals had cerebellar hypoplasia, microcephaly, limb contractures and fetal growth restriction in common. Occurrence of the same mutation in all the five families from India suggested founder effect. The common features with the above two reports were microcephaly, cerebellar hypoplasia, arthogyposis and early neonatal death. Additional findings in our case were an enlarged cavum septum pellucidum and corpus callosal agenesis on ultrasound.
Conclusion
Whole exome sequencing of the affected child identified the specific cause of recurrent congenital CNS malformations. Without this information, the couple would possibly have suffered more losses without an answer. Knowledge about the mutation established the cause, pattern of inheritance and recurrence risk in future pregnancies. It also enabled the maternal fetal medicine team to formulate a definitive care plan for the next pregnancy including early invasive testing and timely ultrasound monitoring. Until state funded facilities enabling access to molecular diagnosis are universal, families and clinicians must be urged to seek a definitive answer by testing the index child.
Learning points
Pontocerebellar Hypoplasia type (PCH) 12 can be diagnosed antenatally by ultrasound scan.
Exome sequencing can confirm the diagnosis by identifying the gene.
In Indian patients with PCH, intronic mutation in COASY gene c.1486-3C>G is a founder mutation which can assist diagnosis by identification of a known pathogenic mutation.
Findings of enlarged fetal cavum septum pellucidum and corpus callosal agenesis in PCH type 12 are novel and require confirmation from further studies.
Disclosure statement: The authors declare that they have no relevant material financial interests that relate to the research described in this paper.
References
- Rudolf G, Lovrečić L, Maver A, Volk M, Peterlin B. Genomic Testing for Prenatal Clinical Evaluation of Congenital Anomalies. Congenital Anomalies - From the Embryo to the Neonate. 2018. http://dx.doi.org/10.5772/intechopen.73247.
- World Health Organization. Global health observatory data. 2015.
- Liu L, Johnson HL, Cousens S, Perin J, Scott S, et al. Global, regional, and national causes of child mortality: An updated systematic analysis for 2010 with time trends since 2000. Lancet. 2012; 379(9832): 2151-2161. doi: 10.1016/S0140-6736(12)60560-1.
- Norman RM. Cerebellar Hypoplasia in Werdnig-Hoffmann Disease: Archives of Disease in Childhood. 1961; 36: 96-101.
- Chou SM, Gilbert EF, Chun RW, Laxova R, Tuffli GA, et al. Infantile olivopontocerebellar atrophy with spinal muscular atrophy (infantile OPCA + SMA). Clinical neuropathology. 1990; 9(1): 21-32.
- Barth PG. Pontocerebellar hypoplasias: an overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993; 15: 411-422.
- Rudnik-Schoneborn S, Sztriha L, Aithala GR, Houge G, Laegreid LM, et al. Extended phenotype of pontocerebellar hypoplasia with infantile spinal muscular atrophy. Am. J Med. Genet. 2003; 117A: 10-17. https://omim.org/entry/607596#.
- Bosveld F, Rana A, Van Der Wouden PE, et al. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum Mol Genet. 2008; 17: 2058-69.
- Srinivasan B, Sibon OCM. Coenzyme A. more than ‘just’ a metabolic cofactor. BiochemSoc Trans. 2014; 42: 1075-9.
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014; 15: 536-50.
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015; 21: 805-21.
- Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, et al. and 15 others. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2014; 94: 11-22.
- Evers C, Seitz A, Assmann B, Opladen T, Karch Set al. Diagnosis of CoPAN by whole exome sequencing: waking up a sleeping tiger’s eye. Am. J. Med. Genet. 2017; 173: 1878-1886.
- van Dijk T, Ferdinandusse S, Ruiter JPN, et al. Biallelic loss of function variants in COASY cause prenatal onset pontocerebellar hypoplasia, microcephaly, and arthrogryposis. Eur J Hum Genet. 2018; 26: 1752-1758. https://doi.org/10.1038/s41431- 018-0233-0.
- Mishra R, Kulshreshtha S, Mandal K, Khurana A, Diego-Álvarez D, et al. COASY related pontocerebellar hypoplasia type 12: A common Indian mutation with expansion of the phenotypic spectrum. American journal of medical genetics. Part A. 2022; 188(8): 2339-2350. https://doi.org/10.1002/ajmg.a.62768.