
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Clinical observations in three clinical cases with locally advanced chordomas. What is needed for early diagnosis with improved survival?
Lena Marinova1* Radoslav Georgiev 2 Nikolay Evgeniev 3
1Complex Oncology Center, Department of Radiotherapy; Russe, Bulgaria
2UMHAT’’St. Marina’’, Varna, Department of Imaging, Radiation therapy and Nuclear medicine; Varna, Bulgaria
3Complex Oncology Center, Department of Medical oncology; Russe, Bulgaria
*Corresponding Author : Lena Marinova
Microbial Biotechnology Department, National Research Centre, Cairo, Egypt
Email: rad_marinova@abv.bg
Received : Dec 15, 2020
Accepted : Dec 30, 2020
Published : Dec 31, 2020
Archived : www.jcimcr.org
Copyright : © Marinova (2020).
Abstract
Chordoma is a rare malignant neoplasm. It is usually diagnosed at an advanced stage, which contributes for the poor prognosis and high mortality. In this article, we will consider three clinical cases with locally advanced chordomas in order to emphasize the importance of early diagnosis by MRI and the need for strict pathohistological and immunohistochemical analysis.
Our observations reported late diagnosis, slow tumor growth, and numerous partial tumor resections, enhancing the mitotic activity of chordoma tumor cells. After numerous operations, rapid local tumor progression with aggressive infiltration of adjacent healthy tissues was observed. Hematogenous dissemination is observed after the third year of primary diagnosis and repeated surgical interventions of locally advanced chordoma. The prognosis in locally advanced chordomas is extremely unfavorable.
Early diagnosis requires MRI and biopsy with exact pathomorphological and immunohistochemical analysis. To improve the treatment results, early diagnosis and complex treatment, including radical surgery and assessment of the need for high-tech postoperative radiotherapy are required.
Keywords: Chordoma, MRI, pathomorphology, immunohistochemical analysis, early diagnosis, surgery, radiotherapy, complex treatment.
Citation: Marinova L, Georgiev R, Evgeniev N. Clinical observations in three clinical cases with locally advanced chordomas. What is needed for early diagnosis with improved survival?. J Clin Images Med Case Rep. 2020; 1(1): 1005.
Background
Chordomas are rare malignant tumors originating from cellular notochordal remnants [1,2]. The most commonly diagnosed chordoma localization is sacral (50-65%), followed by the cranial base and clivus (25-35%) and paravertebral, including cervical (10%) and thoracolumbar (5%) spinal areas [3,4]. Due to the rare incidence, chordoma is diagnosed late, which affects the unfavorable prognosis and high mortality. In this article, we will consider three clinical cases with locally advanced chordomas in order to emphasize the importance of early diagnosis by MRI and the need for strict pathohistological and immunohistochemical analysis.
Three clinical cases with locally advanced chordomas and late diagnosis:
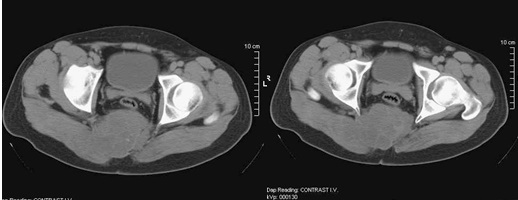
First clinical case with sacral chordoma: A 29 year old man with complaints a gradually increasing painful swelling in the right sacral region in early 2010. He has been assessed with CT which has visualised as a gluteal “cyst” (Figure 1), after that the patient underwent tumor excision with histological result fibromatosis with myxomatous areas.
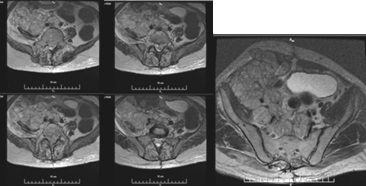
A second operation has been performed in the same year due to persistent pain and growth of the tumor formation in the right sacral region. Chondroid-chordoma has been verified after subtotal extirpation of a soft tissue tumor. Neurological status / 2011- Pain in the right gluteal region, hypoesthesia for sacral dermatomes, controls pelvic reservoirs. Preoperative pelvis MRI / January 2011 (Figure 2) - Sacrococcygeal tumor with osteolysis, bone destruction and expansion; with soft tissue component and dimensions 96 mm (cranio-caudal) / 69 mm (anterior-posterior) / 145 mm (medial-lateral). The formation extends proximally to the sacrum, sacral canal and coccygeal bone; distal-right lateral in the area of the gluteus maximus muscle; retroperitoneally pushes the rectum forward and back - the subcutaneous adipose tissue. Intact rectum with preserved perirectal fat space. The tumor is hypointense on T1 images, hyperintense on T2 images, after contrast slowly increases its inhomogeneity and its signal intensity. Peripherally and centrally with a cellular structure with anintensive septum - cartilaginous tumor matrix. Preserved fatty spaces around the sciatic nerves bilaterally.
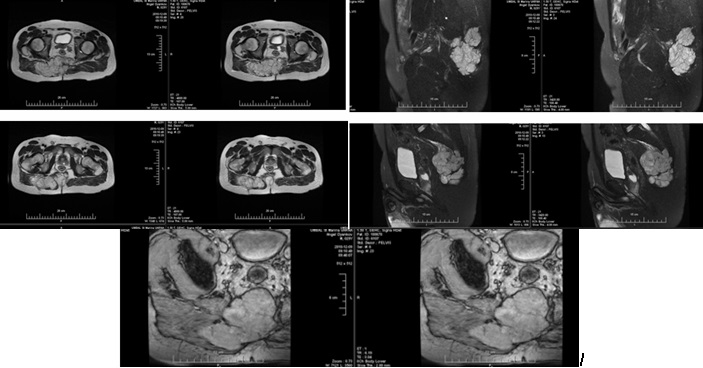
Intraoperatively / February 2011 - A heterogeneous tumor formation in the gluteal mesculature, infiltrating the sacrum and entering the small pelvis with suspicion of infiltration into the rectum has been found. Partial resection of the sacrum with subtotal resection of the soft tissue tumor component has been performed. Postoperative pelvis MRI (Figure 3) /2011- Compared to the transitional study, the tumor dimensions have been reduced: cranio-caudal size 82 mm, anterior-posterior size 47 mm and significant reduction of the transverse size up to 90 mm. Tumor nodules along the right gluteus maximus muscle have persisted. The formation has spread retroperitoneally by pushing forward the rectum, which has been intact - preserved fatty space around it and around the sacral nerves bilaterally.
The patient has been referred for postoperative radiotherapy. Due to a large residual tumor formation with a pronounced pain syndrome, in the Neurosurgical Clinic /University Hospital “St. Ivan Rilski ”Sofia, three more consecutive operations (February /2011; November /2011 and February /2012) have been performed. They have included sacrotomy and subtotal tumor extirpation with visible total excision of the gluteal tumor formation.
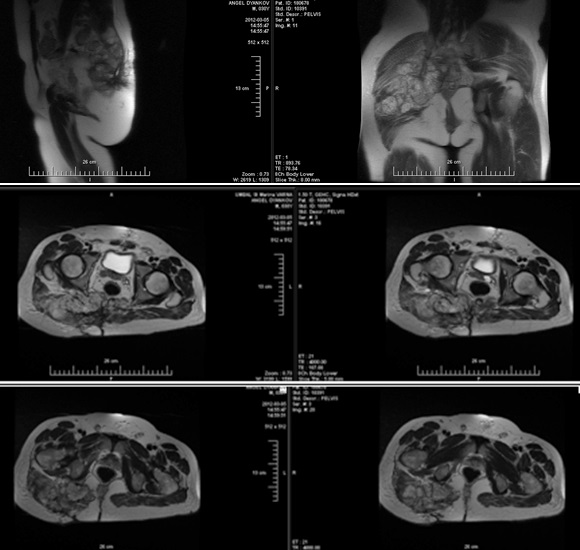
Postoperative pelvis MRI (March /2012) after a month from the fifth operation (Fig.4). Compared to the transient MRI examination, there has been no change in the anteroposterior size and the left component, with significant progression to the right and down, with almost entirely diffuse involvement of the gluteus maximus muscle and into right piriformis muscle. The tumor is hypointense on T1 images and hyperintense on T2 images and after intravenous contrast increases slightly and inhomogeneously its signal intensity. Preserved fatty spaces around the sacral nerves bilaterally. Presence of two enlarged lymph nodes from the right internal obturator circuit with dimensions 17/10 mm and 12/10 mm.
Conclusion: MRI data for residual sacral tumor formation with progression in size and expansion to the right gluteal muscles and to the small pelvis, without involvement of the rectum. Enlarged lymph nodes from right internal obturator circuit. Compared to the previous study, right tumor progression with piriformis muscle growth and involvement of almost the entire right gluteus maximus muscle has been reported.
During the month March/2012, palliative hypofractionated radiotherapy (telegamma therapy) with a clinical target volume (the tumor formation with the surrounding involved structures) with daily dose (DD) 3 Gy upto radiobiologically equivalent total dose (TD) 64 Gy was performed. After radiotherapy, a slight reduction of the pain syndrome was achieved, without visible reduction of the tumor. In September and December/ 2012, two decompression operations (laminectomy of L4 with partial L5 were performed, as well as decompressive chemylaminectomy of L2-3 with partial tumor excision and decompression of the nerve roots) were carried out. December /2012 on MRI a large tumor formation with infiltration of the gluteal muscles on the right was found (Figure 5).

In January 2013 he was admitted again to the Neurosurgical Department with subcutaneously palpable tumor formation in the abdominal area and lower paraplegia. Neurological status: Lower flaccid paraplegia and hypoaesthesia, pronounced pain syndrome in the lumbosacral region. Controls pelvic reservoirs. MRI of the lumbosacral region/January 2013 (Figure 6): Numerous residual tumor formations in the thoracolumbar department, the autochthonous muscles of the spine, abdomen, small pelvis and anterior abdominal wall. Osteolytic soft tissue lesions in the dorsal part of the vertebral bodies Th11 and L2-5, protruding in the spinal canal were visualized. On the Th11 level, the tumor fills almost entirely the spinal canal, as well as the right foramen of Th11-12 with a similar spread at the L2-L5 level. Compression on the nerve roots, more pronounced to the left of the invading tumor formation along the course of the two neuroforamen at the L5-S1 level.
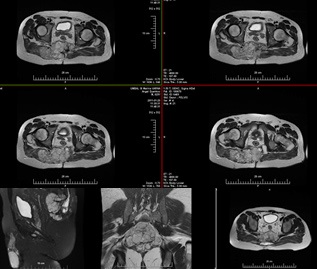
Conclusion: Tumor formation entering the spinal canal from Th10 to the sacrum with direct compression of nerve structures; engaging the small pelvis and abdominal organs. An MRI of the abdomen and pelvis from January 2013 reported a progression of the locally advanced chordoma (Figure 7).
Operation: Under local anesthesia, the spinal canal was exploited with decompression of the right L1 root. An epidural catheter was inserted for analgesic medication. CT of the abdomen and small pelvis / January 2013 (Figure 8): Subpleurally located nodular structures with a solution up to 12 mm. Liver with multiple nodular structures, poorly demarcated, in places confusing up to 40-32 mm. Kidneys - on the right hydronephrosis and hydroureter. Distally to the right, the ureter is traced to the L5 level, where its lumen is lost in an extensive, heterodense lobulated, relatively well demarcated structure, cranially traceable pre- and paravertebral to the right of the L4 level, caudally propagating presacral in the right iliac fossa to the left muscle in the small pelvis. The contours of this structure are well demarcated, dislocating the intestinal loops, bladder, and rectum to the left. Dorsally, this structure erases the contours of the sacrum and coccygeal bone from level S3, propagates to the gluteal muscles dorsally and laterally to the right, propagates through the anterior abdominal wall. Another similar lobulated structure without convincing data for communication with the previous one is established along the line alba / caudally from level L2-3, erasing the contours of muscle rectus avdominis on the right; with axial plan 78/82 mm, caudally traceable to level S3. Numerous nodular structures mesenteric and in the small pelvis with solutions of 27 mm. Numerous osteolytic areas were found in the sacrum, coccygeal axis, right iliac bone, L3-5, and laminectomy in L2, Th11 and Th10. In the portovenous phase, a central defect is found in the lumen of the right femoral vein.
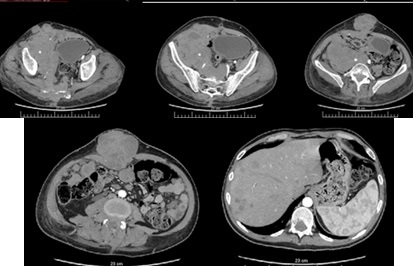
Conclusion: Hematogenous metastatic chordoma in liver, subpleural, bone and mesenteric. Thrombosis of the right femoral vein.
Second clinical case with paravertebral chordoma at the level of TH12-L1: A 75-year-old man with significant comorbidities - hepatitis C, asthma, ankylosing spondylitis, bilateral sacroiliitis, secondary osteoarthritis and osteoporosis. The disease has started with complaints from constant lumbar pain, radiating to the abdomen and lower extremities 2.5 years ago. The patient has been treated for a long period with corticosteroids without effect. With clinical and image data for compression of myelone at the level of Tn12-L1 and has refered for surgical treatment. Partial tumor resection has been performed in 2011 with histological result Malignant neurilemma. Revision of the preparations: Fibrous connective tissue and fragments of cancellous bone with preserved hematopoiesis without tumor infiltration; Tumor tissue composed of polygonal cells with abundant, bright, in some cells vacuolated cytoplasm. The nuclei of tumor cells are oval or irregular in shape with pronounced polymorphism. Part of the nuclei - with protruding nucleoli. Single multinucleated tumor cells as well as cells with large monstrous nuclei. A myxoid matrix is found around groups of tumor cells. Single preserved bone beams. Immunohistochemical analysis: 1 / Cytokeratin (MNF 116): Strong expression in almost all tumor cells ≥ 95%; 2 / Vimentin - moderate to strong expression ≥80% of tumor cells; 3 / S-100 protein - moderate to strong expression ≥60% of tumor cells. Conclusion - Chordoma.
Neurological status / 2011: No symptoms of meningoradicular irritation. Cranial nerves – intact. Pronounced lumbar vertebral syndrome. Lumbo-radicular syndrome with Th12-L1 involvement, more to the left. Lower paraparesis syndrome of central origin. Pelvic tanks - controlled.
From the examinations: MRI of the spine / 2011 - Fracture of the body of Th12 / probably pathological. Pathological mass in the body of L1. CT of the brain - Diffuse small areas with low density - Consequences of a stroke. On 01.08.2011 a laminectomy of Th12 and L1 was performed with foramenotomy bilaterally with transpedicular fixation of Th10-11-L2. Intraoperatively / 2011: Tumor formation, involving the bodies of Th12, L1 and entering the spinal canal, with pronounced compression of the spinal cord, more to the left.
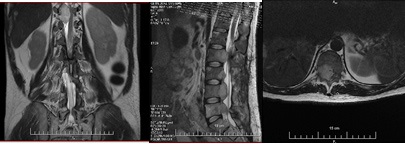
MRI of the spine / December 2012 (Figure 9) - The body of L1 is involved in a pathological lesion with solutions 33/37/48 mm. The lesion has an inhomogeneously high signal intensity of T2 and STIR and low of T1 images. The formation destroys the posterior contours of the vertebral body with expansion to the epidural space and the spinal canal over a wide area. Compression of cone medularis and cauda equina. On T2 and STIR images, the myelone has an increased signal intensity above the affected area - an expression of edema. Neuroforamines at Th12-L1-L2 levels are obliterated bilaterally. The body of Th12 is wedge-shaped lowered with inhomogeneously high signal intensity of STIR and low of T1 images, as an expression of edema. The bodies of the other vertebrae have diffusely inhomogeneous bone marrow signal intensity with high signal foci in T1 and T2, probably an expression of inhomogeneous fat transformation. Subchondral linear focus persists on the lower disc surface of L2 with high signal intensity of T2 and low of T1 - as in bone marrow edema. MRI data for a tumor lesion of the body of L1 with expansion to the spinal canal and pathological fracture of Th12.
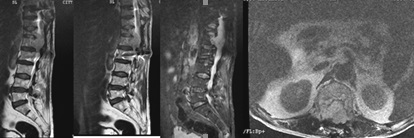
The patient has been stabilized for several months, after which the pain intensifies. Local neurological status /2013- Pain vertebral syndrome of the lumbar segment. Lower paraparesis. No symptoms of meningoradicular irritation. Cranial nerves - intact; Pelvic reservoirs - urinary incontinence. The patient has been refered for reoperation after MRI of the spine in April 2013. Intraoperatively from the second operation The dura mater of the spinal cord was reached. A tumor formation on both sides of the dura was seen. It was partially removed.
Third clinical case with lumbosacral chord in a 25-yearold girl: Aproximately 1 year history for pain syndrome in the sacral region, accompanied by tingling on the posterior surface of the left thigh. After MRI and biopsy, the pathohistological diagnosis have been significantly uncertain- chordoma or alveolar rhabdomyoblastoma? The patient has been referred to a neurosurgery clinic for maximal cytoreduction.
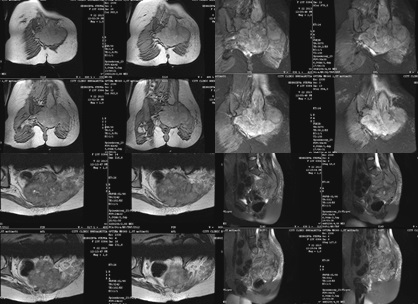
From the studies: Preoperative MRI (Fig. 10) - A large tumor formation involving the sacrum, pushing the rectum forward, partially involving the left iliac bone.
Neurological status - No symptoms of meningoradicular irritation. Cranial nerves - intact; Without paresis or paralysis, chronic pain in the plexus lumbosacralis. Sensory disturbances in the perineum and left gluteal region. Retention of pelvic reservoirs. Local status of the left gluteal region - fresh surgical scar after biopsy. A large formation with a diameter of about 20 cm is palpated. with significant prominence. First operation / June 2013 Intraoperatively: A tumor formation invading the sacral bone was found, mainly the left half, with a mushy crumbly characteristic. The sacrum is destroyed. The bony edges of the rest of the sacrum are palpated. The tumor was aspirated and excised as much as possible. Heavily bleeding from tumor formation was the reason for interruption of the surgical intervention. The macroscopic characteristics of the tumor resemble a chordoma. The tumor formation is partially extirpated. Histological result / June 2013 - Chordoma.
Postoperatively status, self-defecation was restored pain was relieved. Urinary retention persists. Independent gait is possible. Over a period of one month, the pain progressed, radiating to the right leg and immobilizing it. After embolization of the feeding blood vessels, reoperation was considered. Local status of the left gluteal region - The skin above the gluteal region is stretched and bombarded by an extremely large tumor formation. Dehiscence in the right corner of the operative scar. Neurological status: Meningoradicular irritation not detected; Cranial nerves - no changes. Decreased in volume and strength movements in the lower extremities, chronic pain in the plexus lumbosacralis - mostly on the left. The patient can control motorcycle fuel tanks. Preoperative MRI - А large tumor formation involving the sacrum, pushing the rectum forward, involving partially the left iliac bone to the S2 vertebra inclusive. Intraoperatively from the second operation / August 2013- A strongly grown tumor with violet color was found under the skin, infiltrating the gluteal muscles with prolapse above it. A large tumor mass with many necrotic areas and heavy bleeding is excised. Postoperative neurological status - Lower severe paraparesis with impaired sensation without controlling the pelvic reservoirs.
Discussion
Chordoma is a rarely diagnosed borderline malignant tumor. As it is an extremely radiation- and chemoresistant neoplasm, early diagnosis, including MRI [5-7] and biopsy with strict pathohistological and immunohistochemical analysis [1,8-10] is of great importance for complex treatment. The prognosis is directly related to the possibility of radical surgery, the residual postoperative tumor volume, the pathohistological characteristics (histological variant and the presence of tumor necrosis), and the sex of the patient [8,9,11,12]. Of the presented three clinical cases with locally advanced chordoma, late diagnosis, difficult pathohistological differential diagnosis (DD) and radical surgery depending on the tumor volume and location are reported. The listed characteristics are significant factors worsening the prognosis of chordoma disease. The discussion highlights the following important aspects:
Difficult pathohistological differential diagnosis (DD) - In the first clinical case, “fibromatosis” has been diagnosed, and after an interval of almost 2 years, a late diagnosis of chondroid / chordoma was made. In the second clinical case, the initial pathohistological diagnosis has been malignant neurilemma, and only after revision with immunohistochemical (IHC) analysis, the final diagnosis of chordoma has been proved. There are three different subtypes of chordoma, namely, chondroid, dedifferentiated, and classical, with the chondroid type having better prognosis than the classical type [13]. Three pathohistological variants are reported: spindleshaped, chondroid or epithelioid, extremely rarely rhabdoid [7-9,14]. Chondroid chordoma is originally described as a variant of chordoma that contained cartilaginous areas indistinguishable from hyaline type chondrosarcoma, and they have better prognosis than non-chondroid chordomas [13]. The histological appearance of classical chordoma is of a lobulated tumor composed of groups of cells separated by fibrous septa. The cells have small round nuclei and abundant vacuolated cytoplasm, sometimes described as physaliferous (having bubbles or vacuoles) [15]. The following histologic parameters have to be evaluated: tumor matrix (chondroid or myxoid [non-chondroid]), growth pattern (lobulating or diffuse), necrosis, presence and proportion of benign notochordal cell tumor (BNCT)-like component (sheets of large uni- or multivacuolated adipocyte-like cells with eccentrically located small nuclei and lacked the intervening tumor matrix), nuclear pleomorphism, and intracytoplasmic hyaline globules [16]. There are few clinical cases published with rare anaplastic undifferentiated hormones with participants in highly malignant sarcoma [7,9]. Exact pathohistological diagnosis requires strict IHC analysis, taking into account the constellation for notochordal tumor chordoma. By IHC, the tumor cells are positive for CK, AE1/AE3, S-100 protein, and INI1/SMARCB1 [17]. The best immunohistochemical markers useful for the evaluation of tumors with chordoid morphology are D2-40, EMA, cytokeratin, and GFAP. D2-40 is a true chondroid marker to be useful for the differential diagnosis with chordoma [18]. Of the chordomas presented, only in the second clinical case was IHC performed with the following positive expression: 1 / Cytokeratin (MNF 116)- Strong expression in almost all tumor cells ≥ 95%; 2 / Vimentin - moderate to strong expression ≥80% of tumor cells; 3 / S-100 protein - moderate to strong expression ≥60% of tumor cells. The differential diagnosis of chordoma includes: 1 / Metastasis of clear cell carcinoma; 2 / Chondrosarcoma - variant of choroidal sarcoma; 3 / Anaplastic chordoma imposes DD with spindle-shaped alveolar rhabdomyosarcoma (in children and young adults) and anaplastic undifferentiated sarcoma.
Characteristic chordoma MRT image - In chordomas a characteristic variable MR signal of T1 images is reported. The tumor spread in the bone marrow substance leads to a reduced intensity of the bone marrow MR signal. In T2 images, most tumors show a hyperintense signal on T2 images, with possible variability depending on the presence of cysts, solid components, and hemorrhages (Figure 2, Figure 4). The increased signal of T1 images is due to hemorrhagic areas and areas with mucosal or protein content. The areas of reduced MR signaling of T2 images are due to calcifications, bone sequesters and fibrous septa. In STIR mode, a hyperintense signal is reported [5,7].
Typical biological development of chordoma - In the presented three clinical cases with locally advanced chordomas, we observe a slow tumor growth with local infiltration in the neighboring tissues and organs. Although considered to be a low-grade and slow-growing tumor, a poor long-term prognosis is generally observed due to extensive local recurrences and secondary complications [19].
Each partial resection is followed by aggressive tumor proliferation as a result of improved tumor blood supply and increased mitotic index of tumor cells [10,20]. In the first clinical case of sacral chordoma, a three-year tumor growth was observed against the background of repeated operations, followed by hematogenous metastases - liver, subpleural and mesenteric. Similar late distant metastases have been published in the literature [19]. In general, the chordoma is rich in blood supply, with a high risk of massive intraoperative bleeding [10]. In the first operation of the third clinical case, heavy bleeding was observed, which necessitated its cessation.
Multiple surgical interventions - In the first clinical case, five partial operations heve been performed for a period of two years, followed by three palliative / decompressive chemylaminectomies. The large tumor volume requires resection through the tumor, followed by macroscopic tumor remnants - R1-2 resections. Even in small chordomas. Due to the immediate tumor proximity to vital brain centers and structures, as well as tumor infiltration of nerve plexuses, cavities or parenchymal organs, maximum radical surgery is possible in 50% of patients, even in small chordomas [16,21,22]. Аlthough surgical approach and total tumor resection are difficult in skull base chordomas (SBCs) patients, residual tumors were not an independent risk factor of recurrence in multivariate analysis [16].
The optimal treatment of chordoma includes maximally radical surgery, followed by high-tech radiotherapy (RT) [20,23,24]. Surgery is the mainstay of treatment and local control is better achieved with en bloc resection of the tumor with safe margins [19,25- 27]. Patients who underwent resections with negative margins showed a better prognosis then those with positive margins [28]. Radiotherapy is generally used only as adjuvant treatment after incomplete surgical resection, or in palliative management [25]. York et al. have reported prolonged progression-free survival with radiotherapy for patients with subtotal resection [29]. In small chordomas, radiosurgery with stereotactic high-tech radiotherapy equipment Gamma-Knife or modified accelerator (Cyber-Knife) with realization of high single radiation doses is applicable with very good healing results [30,31]. Radiosurgery is not suitable for advanced, large-volume tumors located near critical structures such as the brain chiasm, optic nerves, and brainstem due to the high risk of late radiation toxicity [1,3,12,32,33]. The pronounced chordoma radiation resistance imposes RT with accelerated highenergy protons, accelerated helium (He) and hydrogen (C12) ions [1,20,24]. The aim is to improve local tumor control by realizing high carcinogenic doses while maximally sparing the surrounding normal tissues [20,23,33]. After combined treatment, including surgery and postoperative stereotactic RT with DD 2-2.4 Gy up to TD 70-84.2 Gy, good treatment results 63% 5-year disease-free survival were reported [12,32,33]. In combined photonic and proton RT after realization of TD 66-83Gy with DD 2 Gy, 73% 5-year disease-free survival was achieved [24]. In inoperable chordomas, LL is palliative, because despite the realized high radiobiologically equivalent TD 64Gy, temporary analgesia is observed without significant reduction of tumor volume [10]. The palliative analgesic effect of RT is not a consequence of cell death of the large tumor, but a result of anti-inflammatory and decompressive effect on the structures adjacent to the tumor.
Conclusion
Chordoma is a rarely diagnosed malignant neoplasm. The main treatment is maximally radical surgery, which depends on the tumor location and is possible in 50% of patients. Despite the known chordoma radiation resistance, radiosurgery and accelerated proton radiotherapy are alternative treatments for small chordomas. Our observations reported late diagnosis, slow tumor growth, and numerous partial tumor resections, enhancing the mitotic activity of tumor cells. After numerous operations, rapid local tumor progression was observed with aggressive infiltration of adjacent healthy tissues. Hematogenous dissemination is observed after the third year of primary diagnosis and repeated surgical interventions of locally advanced chordoma. The prognosis in locally advanced chordomas is extremely unfavorable. Early diagnosis requires MRI and biopsy with exact pathomorphological and immunohistochemical analysis. To improve the treatment results, early diagnosis and complex treatment are required, including maximum radical surgery and assessment of the need for high-tech postoperative RT.
References
- Marinova L, Vasileva V. Radiation therapy of benign diseases. Galakta Publishing House. 2010; 73-75.
- Walcott BP, Nahed BV, Mohyeldin A, Coumans J-V, Kahle KT, Ferreira MJ. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012; 13: e69-e76.
- Nguyen RP, Salzman KL, Stambuk HE, Ahuja AT, Harnsberger HR. Extraosseous Chordoma of the Nasopharynx American Journal of Neuroradiology. 2009; 30: 803-807.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System Geneva: World Health Organization. 2016.
- Gehanne C, Delpierre N, Damry N, Devroede B, Brihaye P, Christophe C. Skull base chordoma: CT and MRI features. JBR-BTR. 2005; 88: 325-327.
- Anegawa T, Rai M, Hara K, Yamamoto K, Narumi O, Hashimoto K, et al. An unusual cervical chordoma: CT and MRI. Neuroradiology. 1996; 38: 466-467.
- Sirodkin DV. Chordomy of the skull base. Methods of surgical and combined treatment. Abstract of the thesis. Dissertation of Doctor of Medical Sciences. 2009; 22.
- Al-Adnani M, Canon SR, Flanagan AM. Chordomas do not express CD10 and Renal Carcinoma (RCC) antigen: an immunohistochemical study. Histopathology. 2005; 47: 535-537.
- Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of Skull Chordomas in Children and Adolescents: A Clinicopathologic Study of 73 cases. Am. J Surg Pathol. 2006; 30: 811-818.
- Marinova L. Radiotherapy in the complex treatment of primitive germ cell tumors. Dissertation of Doctor of Medical Sciences. 2013; 36-38.
- Weber AL, Liebsch NL, Sanchez R, Sweriduk ST. Chordoma at the skull base. Neuroimaging Clin N Am; 1994; 4: 515-527.
- Sirodkin DV, Makhmudov UB, Shimansky VN. Cranial chordoma. Localization, surgical methods of treatment. Prognostic factors i. International Congress of Oncosurgery, Oncosurgery. 2008; 2: 99.
- Heffelfinger MJ, Dahlin DC, MacCarty CS, Beabout JW. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973; 32: 410- 420.
- Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma. Am J Surg Pathol. 1998; 22: 1083- 1092.
- Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: the nonsarcoma primary bone tumor. The Oncologist. 2007; 12: 1344-1350.
- Cha YJ, Suh Y-L. Chordomas: Histopathological Study in View of Anatomical Location. J Korean Med Sci. 2019; 34: e107.
- Rekhi B, Banerjee D, Ramadwar M, Bajpai J, Jambhekar NA. Clinicopathologic features of four rare types of chordomas, confirmed by brachyury immunostaining. Indian J of Pathology Microbiology. 2017; 60: 350-354.
- Cho H-Y, Lee M, Takei H, Dancer J, Ro JY, Zhai AJ. Immunohistochemical comparison of chordoma with chondrosarcoma, myxopapillary ependymoma, and chordoid meningioma. Appl Immunohistochem Mol Morphol. 2009; 17: 131-138.
- Fourney DR, Gokaslan ZL. Current management of sacral chordoma. Neurosurg Focus. 2003; 15: E9.
- Marinova L, Hadjieva T. Manual of Radiobiology; Galakta Publishing House. 2009; 61.
- Gulyaev D. Surgery for tumors of the base of the cranial fossa. Author’s abstract. dissertation of Dr Med Sciences. 2011; 20.
- Tanyashin SV. Surgical aspects of the treatment of malignant tumors affecting the base of the skull: Author’s abstract. Dissertation of Doctor of Medical Sciences. 2005; 15.
- Marinova L, Yaneva M. Guide to radiotherapy for students. Galakta Publishing House. 2008; 81.
- Feuvret L, Noel G. La proton therapy une technique d irradiation meconnue. 2003; 53: 701-703.
- Baratti D, Gronchi A, Pennacchioli E, Lozza L, Colecchia M, Fiore M, et al. Chordoma: natural history and results in 28 patients treated at a single institution. Ann Surg Oncol. 2003; 10: 291-296.
- Bergh P, Kindblom LG, Gunterberg B, Remotti F, Ryd W, MeisKindblom JM. Prognostic factors in chordoma of the sacrum and mobile spine: a study of 39 patients. Cancer. 2000; 88: 2122- 2134.
- Lopes A, Rossi BM, Silveira CR, Alves AC. Chordoma: retrospective analysis of 24 cases. Sao Paulo Med J. 1996; 114:1312-1316.
- Junior SA, Andrade WP, Baiocchi G, Guimaraes GC, Cunha IW, Estrada DA, et al. Natural history and surgical treatment of chordoma: a retrospective cohort study. Sao Paulo Med J. 2014; 132: 297-302.
- York JE, Kaczaraj A, Abi-Said D, Fuller GN, Skibber JM, Janjan NA, et al. Sacral chordoma: 40-year experience at a major cancer center. Neurosurgery. 1999; 44: 74-79.
- Wackym P. Stereotactic Radiosurgery, Microsurgery, and Expectant Management of Acoustic Neuroma: Basis for Informed Consent. Otolaryngologic Clinics of North America. 2005; 38: 653-670.
- Benovic R, Zivkovic N, Stanic M. Svanomi i meningeomi- najcesci tumori PCU I njihov klinicki znacaj. Materia medica. 2011; 27: 217- 220.
- Sirodkin DV, Makhmudov UB, Golanov AV. Combined treatment of cranial chordomas. Surgical access i. Modern methods of radiation therapy. International Congress of Oncosurgery, Oncosurgery. 2008; 2: 99.
- Sirodkin DV, Golanov AV, Makhmudov UB. Stereotactic radiotherapy with chordomas of the skull base. V Congress of Neurosurgeons of Russia: Materials of the Congress. Publishing house “Health of Bashkortostan”. 2009; 302.