
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Treatment of coronavirus disease: Implementation of machine learning algorithms for drug screening
Baez MDC1; Scribano Parada MP1; Picco Berrotarán FM1; Rossi MM1; Marchi AA2; De Francesca LA2; Castillo TA1; Balceda AGA1
1 Cathedra of Biomedical Physics, Faculty of Medical Sciences, National University of Córdoba, Argentina.
2 Faculty of Mathematics, Astronomy and Physics, National University of Córdoba, Argentina.
*Corresponding Author : María del Carmen Baez
Cátedra de Física Biomédica, Facultad de Ciencias
Médicas, Universidad Nacional de Córdoba, Santa
Rosa 1085, cp-5000. Córdoba, Argentina.
Email: baezmdc@yahoo.com.ar
Received : Feb 15, 2021
Accepted : Mar 11, 2021
Published : Mar 15, 2021
Archived : www.jcimcr.org
Copyright : © Baez MC (2021).
Abstract
Background and objectives: The pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) led to the emergence of the immediate and urgent need to develop of therapeutic measure capable of reducing its impact on the health of the population and in health and economic resources. Based on the data provided by these bioassays, in this work propose the implementation of machine learning algorithms based on a Quantitative Structure-Activity Relationship (QSAR) model for drug screening of compounds with the potential to inhibit Quinone Reductase 2 (QR2) and to replace the anti-inflammatory function of chloroquine and hydroxychloroquine in the treatment of COVID-19 avoiding its adverse effects.
Methods: QSAR modeling was performed to calculate the mathematical correlations between the chemical properties of QR2 inhibitor compounds, from different bioassays, and their biochemical response on QR2 activity. The values of 22 properties were obtained by means of automatic extraction techniques from PubChem’s PUG REST service. The following classification algorithms were applied: Logistic Regression, Random Forest and Multi-Layer Perceptron. To perform the computational screening, 279 drugs were selected and divided into 7 groups: Group I or PubChem-Covid-19, settled for compounds labeled by PubChem as COVID-19 (n=104); Group II, drugs with structure similar to dihydroxyphenylalanine (dopa) (n=110); Group III, ubiquins (n=16); Group IV, used in clinical trials (n=18); Group V, amantadine, pramipexole, dabigatran, rotigotine and naphthoquinone (n=5); Group VI, vitamins B (n=10); and Group VII, vitamins K (n=16). A classification threshold for Active of 0.95 was established.
Results: 54 compounds were identified as Actives. Camostat, relacatib, 5-Aminopyrimidine, clovamide, coenzyme Q4, decylubiquinone, sarilumab, fingolimod, rivaroxabán, prosultiamine and alinamin, for its potential use in COVID-19, were the most significant.
Conclusions: It was presented a series of compounds identified by the QSAR model as QR2 inhibitors and we analyze the main drugs in that series according to their availability and current use.
Keywords: COVID-19, Chloroquine, Hydroxychloroquine: Quinone reductase 2, Machine learning, QSAR model.
Citation: Baez MDC, Scribano Parada MP, Picco Berrotarán FM, Rossi MM, Marchi AA, et al. Treatment of coronavirus disease: Implementation of machine learning algorithms for drug screening. J Clin Images Med Case Rep. 2021; 2(2): 1025.
Introduction
The pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), declared in March 2020 by the WHO, led to the emergence of the immediate and urgent need to understand and investigate the different mechanisms of infection of this virus for the development of therapeutic measures capable of reducing its impact on the health of the population and in health and economic resources.
SARS-CoV-2 can cause from mild infection to Acute Respiratory Distress Syndrome (ARDS) causing 5 to 12% of patients to require admission to the Intensive Care Unit (ICU) [1]. It is known that SARS-CoV-2 is caused by a β coronavirus, consisting of a singlechain linear RNA and positive polarity. Its genetic material has a 5´methylated head and a 3´ polyadenylated tail, encoding proteins in the sense of 5´ to 3´ which generates similarity with the host mRNA giving it the possibility of adhering to the ribosomes for direct translation into the 1a/1ab polyprotein inducing the infection [2]. In addition, this virus has in its external structure an S glycoprotein that is activated by a specific enzyme called furin in the host cells, which mediates the binding to the AngiotensinConverting Enzyme 2 (ACE2) receptor. This binding has a force at least ten times greater than the S protein from severe acute respiratory syndrome coronavirus (SARS-CoV), and it could be key point in SARS-CoV-2 infection [3-5]. This would explain, among other characteristics, the high rate of infected people. In turn, the wide distribution of the enzyme furin in different tissues (liver, lung and small intestine) would also explain the multiorgan attack of this virus [6].
So far, the implementation of Chloroquine (CQ) and Hydroxychloroquine (HCQ) has been one of the most tested therapeutic alternatives against SARS-CoV-2 due, in particular, to the ability of CQ to inhibit other coronaviruses [7]. CQ/HCQ are quinolins, heterocyclic aromatic compound containing nitrogen. Quinoline ring in these compounds has been shown to possess antimalarial, antibacterial, antifungal, anthelmintic, cardiotonic, anticonvulsant, anti-inflammatory and analgesic activity [8]. In recent in vitro studies, CQ was found to be highly effective in controlling SARSCoV-2 infection and its evaluation has been suggested in human patients suffering from the new coronavirus disease [9]. Although some clinical trials have shown that CQ/HCQ could inhibit the exacerbation of pneumonia and promote negative virus conversion by shortening the disease [10]. There were conflicting preliminary clinical data in others [11]. In addition, along with other adverse effects, CQ/HCQ can generate life-threatening cardiac arrhythmias by interfering with ventricular repolarization by prolonging the QTc interval causing torsade de pointes (TdP) [12]. Because of this is that it is suggested to use CQ/HCQ in carefully selected and monitored patients [13].
The evaluation of the antiviral effects of CQ/HCQ in vitro against SARS-CoV-2 infection suggests that they could inhibit the entry of the virus into the cell, causing a deficiency in glycosylation of the ACE2 receptor [7]. They would also affect viral replication after cell admission by affecting the endosomal–lysosomal interaction [14,15]. However, the biological targets of CQ and HCQ are few known. In 2002, Graves et al., demonstrated that the enzyme quinone reductase 2 (QR2) is a cytosolic flavoenzyme that uses FAD as a substrate and dihydronicotinamide riboside (NRH) as a reducing coenzyme, is a selective target of quinolins, including CQ/HCQ, and that they strongly inhibit their activity [16]. Subsequently, Kwiek et al., suggested on the effects of quinolins in vivo and recommended studies aimed at understanding the physiological importance of QR2 and creating inhibitors of this enzyme [17]. The QR2 gene is mainly expressed in kidney, liver and heart. In red blood cells, the QR2 gene is expressed intermediately while in the brain and pancreas its expression is minimal [18]. QR2 inhibition is known to be associated with positive regulation of antioxidant enzymes, although the mechanisms have not been fully elucidated. The antioxidant activity of melatonin and resveratrol reported in numerous studies could be due to the fact that these drugs are potent QR2 inhibitors [19,20]. Currently, strong evidence supports those observations. Gould et al., have recently shown that an increase in QR2 activity causes the generation of Reactive Oxygen Species (ROS) and that the reduction of its expression, therefore, is a way of reducing inflammation, therefore QR2 can be considered a redox modulator [21].
The beneficial effects of CQ/HCQ on Coronavirus Disease 2019 (COVID-19) could be due not only to its antiviral effects, but also to its anti-inflammatory effect and its ability to inhibit QR2. Furthermore, if the evidence provided by critically ill patients with SARS-CoV-2 is taken into account, they have an elevated cytokine profile, similar to that observed in Cytokine Storm Syndrome (CS) in SARS and MERS [22]. Extremely high inflammatory parameters such as C Reactive Protein (CRP) and pro-inflammatory cytokines (IL-6, TNFα, IL-8) are observed. Clinically expressed with vasculitis, hypercoagulability and multi-organ damage. This is why immunologists consider that applying a timely anti-inflammatory treatment, adapted to each patient, is of vital importance in COVID-19 [23].
Despite the dual capacity of CQ/HCQ, inhibit viral replication by different mechanisms and possess anti-inflammatory action, its implementation has limitations due to adverse effects [24]. Therefore, an expert panel that analyzed the use of CQ/HCQ in SARS-CoV-2, recommended several precautions, including blood tests to discard the development of anemia, thrombocytopenia, leukopenia, electrolyte disorders, liver dysfunction, and kidney dysfunction. It was also recommended serial electrocardiography to exclude prolongation of the QTc interval and regular interviews for the early detection of visual disturbances and mental state, although the latter usually occur with long-term use of drugs [25]. Therefore, the use of drugs capable of mimicking CQ/HCQ could be a significant treatment alternative. As QR2 is one of the few well-known and well-defined biological targets of CQ and HCQ, other drugs capable of inhibiting its function could replace them or be adjuvants in the treatment of SARS-CoV-2 infection when administered as concomitant treatment with CQ or HCQ. This would allow to reduce the dose of these, with it the appearance of dose-dependent effects and/or to prolong the treatment with them.
Given the global urgency of the pandemic regarding the need for therapeutic resources, the Quantitative Structure-Activity Relationship (QSAR) computational models could be a valuable resource as a tool for drug screening, immediately and automatically, for the detection of drugs capable of imitating the effects of CQ/HCQ [26,27]. There are currently numerous bioassays that evaluate the inhibition of human QR2 by numerous compounds. They provide valuable data for computational analysis and prediction of effects on QR2 by drugs that were never specifically tested on QR2 activity. These bioassays have been carried out in the last decade based on the fact that the inhibition of QR2 leads to the protection of cells against ROS [28]. Based on the data provided by these bioassays, in this work we propose the implementation of machine learning algorithms based on a QSAR model for drug screening of compounds with the potential to inhibit QR2 and to replace the anti-inflammatory function of CQ and HCQ in the treatment of Coronavirus disease avoiding its adverse effects.
Methods
QSAR modeling was performed to calculate the mathematical correlations between the chemical properties of QR2 inhibitor compounds, from different bioassays, and their biochemical response on QR2 activity. The biochemical response of the drugs was defined as a function of the chemical properties of the bioassayed compounds [29].
Biochemical response =f(chemical properties of bioassayeddrugs)
The basic steps performed for QSAR modeling were: (i) data selection; (ii) data preparation; (iii) data processing; (iv) data prediction and validation; and (v) data interpretation. During data preparation and validation, multiple test instances were performed. In each step of the QSAR model, various statistical operations were involved.
Bioassays
A total of 61 bioassays available in PubChem were analyzed for the search “quinone reductase 2”, from which 18 bioassays were selected. The main selection criterion was the inhibition of the enzymatic activity of human QR2. Those bioassays on non-human QR2 or in which the Target Name was different, were rejected.
Selected bioassays included chemopreventive compounds, with known QR2 inhibitory activity, casimiroin [30], resveratrol [31,32], phenylethanoid glycosides [33], MCA-NAT (5-methoxycarbonylamino-N-acetyltryptamine) [34], phenazine [35] and ammosamide B [31].
A bioassay was also included in which QSARs models were developed using ML techniques, for derivatives of naphthalene, benzofurane and indole with respect to their affinities with the melatonin binding site in QR2 [36].
Datasets and properties
156 compounds were obtained from the bioassays after eliminating those repeated and/or without data. Of the compounds obtained, 123 were reported as Active (QR2 inhibitors). Compounds reported as inactive, inconclusive or unspecified were considered as non-inhibitors of QR2. Finally, the dataset was balanced up to 256 compounds to equal the number of Actives respect to the number of Inactives. To this end, compounds with a structure similar to those reported as Inactive were randomly selected and added to the database, with no evidence of activity against QR2.
The values of 22 properties, for each one of the compounds, were obtained by means of automatic extraction techniques from PubChem’s PUG REST service (Table 1).
Table 1: List of properties ordered from highest to lowest according to the coefficient assigned by the Logistic Regression model.
Coefficient |
||
1 |
Number of atoms with defined planar (sp2) stereo |
1,725,880 |
2 |
Number of bonds with planar (sp2) stereo |
1,542,999 |
3 |
Number of rings |
1,024,749 |
4 |
Conformer sampling RMSD in Å |
0,685816 |
5 |
Number of anionic centers (at pH 7) |
0,478884 |
6 |
Feature count 3D |
0,313042 |
7 |
Number of hydrophobes |
0,256168 |
8 |
Number of hidrogené-bond acceptors in the structure |
0,206096 |
9 |
Number of rotatable bonds |
0,204355 |
10 |
Effective rotor count 3D |
0,172259 |
11 |
Y steric quadrupole 3D |
0,064420 |
12 |
Topological polar surface area (TPSA) |
0,039569 |
13 |
Molecular complejita |
-0,000893 |
14 |
X steric quadrupole 3D |
-0,033168 |
15 |
Number of conformers |
-0,088578 |
16 |
Number of non-hydrogen atoms |
-0,099296 |
17 |
Number of cationic centers (at pH 7) |
-0,122491 |
18 |
Computationally generated octanol-water partition coefficient or distribution coefficient (XLogP) |
-0,162769 |
19 |
Number of hydrogen-bond donors of a conformer |
-0,284483 |
20 |
Number of hydrogen-bond donors in the structure |
-0,678290 |
21 |
Z steric quadrupole 3D |
-0,789037 |
22 |
Number of hydrogen-bond acceptors of a conformer |
-1,039786 |
A second set of test data was made up of 24 Active compounds and 62 Inactive compounds for the evaluation of the models with unknown data.
Selection of characteristics and classification algorithms
The following classification algorithms were applied: Logistic Regression, Random Forest and Multi-Layer Perceptron. All algorithms were applied in the Python programming environment, v. 3.7. The scikit-learn library, v. 0.22.2 was used to model the algorithms. Data manipulation and analysis was performed with the Numpy and Pandas libraries, and the visualization of the same with Matplotlib and Seaborn. Scikit-learn’s Feature selection module was used for property selection.
The evaluation of the estimator’s performance was performed by cross-validation. The grid search technique was used to find the optimal hyperparameters of the algorithms.
Drugs
To perform the computational screening, 279 drugs were selected according to: their structural chemical similarity with the bioassayed compounds; its potential action on QR2; or for use in patients with COVID-19. They were divided into 7 groups. One of those groups, Group I or PubChem-Covid-19, settled for compounds labeled by PubChem as COVID-19 (n=104). The other groups were formed as follows: Group II with drugs with a chemical structure similar to dihydroxyphenylalanine (dopa) (n=110); Group III with drugs of chemical structure similar to ubiquins (n=16); Group IV made up of drugs with drugs used in clinical trials, or evaluated for possible use, such as azithromycin, baricitinib or bevacizumab, among others (n=18); Group V with amantadine, pramipexole, dabigatran, rotigotine and naphthoquinone (n=5); Group VI vitamins B (n=10); and Group VII vitamins K (n=16).
Classification threshold
A classification threshold for Active of 0.95 was established.
Results
Model validation and test with unknown data
During the model validation stage, the Logistic Regression algorithm obtained the best performance: accuracy 88.4%; precision 88.4%; and recall 88.4%. Equality between the accuracy, precision and recall metrics means that the algorithm classified an equal number of positive and negative cases, that is, the training dataset correctly balanced between positive and negative cases. For the Logistic Regression model, the data set was partitioned into 80% for training and 20% for testing. Figure 1 and Figure 2 shows the confusion matrices of the tests of the Logistic Regression model with unknown data.
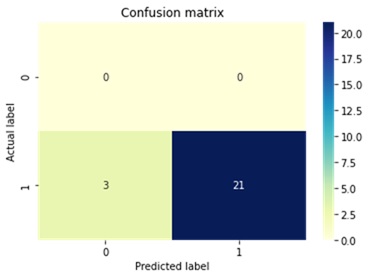
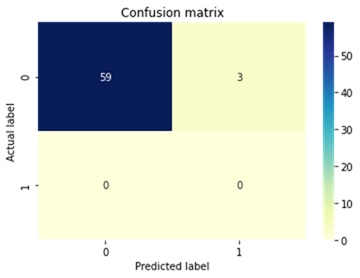
Selection of properties
By using the Feature selection module, reducing the number of properties showed a tendency to classify most compounds as Active, so we decided to use all available features to supply more information to the model. Table 2 shows the coefficient assigned by the Logistic Regression model to all properties.
Table 2: Most relevant drugs detected by group, ordered from highest to lowest according to their probability of inhibiting QR2.
Group I (n=104) |
Classification threshold |
Camostat |
0.99688 |
Relacatib |
0.98327 |
5-Aminopyrimidine |
0.95696 |
Classification threshold |
|
N-Caffeoyl DOPA (Clovamide) |
0.99255 |
Group III (n=16) |
Classification threshold |
Coenzyme Q4 |
0.99997 |
Ubiquinone Q3 |
0.99966 |
A2-Ubiquinone-2 |
0.99810 |
Decylubiquinone |
0.99743 |
A6-Ubiquinone-2 |
0.96759 |
Group IV (n=18) |
Classification threshold |
Sarilumab |
0.99280 |
Fingolimod |
0.99141 |
Rivaroxaban |
0.97753 |
Classification threshold |
|
Amantadine |
0.99152 |
Pramipexole |
0.97042 |
Group VI (n=10) |
Classification threshold |
Prosultiamine |
0.99994 |
Alinamin |
0.99975 |
Group VII (n=16) |
Classification threshold |
Menatetrenone |
0.99997 |
Vitamin K2 |
0.99586 |
Phytonadiol |
0.99585 |
0.99056 |
|
Phytonadione |
0.99056 |
Vitamin K |
0.95642 |
Virtual screening
A total of 54 compounds were identified as Actives. Table 2 shows the most relevant drugs detected by group, ordered according to their probability value from highest to lowest.
In Group I or PubChem-Covid-19, of 104 compounds, 7 were labeled as Active. In the case of Group II, drugs with a dopa-like chemical structure, out of a total of 110 compounds, 23 were recognized as active by the algorithm. In Group III, of 16 compounds with a chemical structure similar to ubiquins, 5 were Active, while in Group IV (clinical trials), only 3 of 18 drugs were labeled in this category. In Group V, only amantadine and pramipexole exceeded the threshold. In the case of the group B vitamins, Group VI, of 10 compounds they exceeded the threshold only 2. Finally, vitamins K, Group VII, 11 of 16 were Active.
Discussion
In this work, we use ML techniques for the development of a binary classification QSAR model to predict the inhibitory activity of QR2 by untested compounds. Verify the model following the best QSAR modeling practices [37], complying with the guidance of the Organization for Economic Cooperation and Development (OECD) [37].
QR2 was strategically selected as a pharmacological target, as it would be a common point at the crossroads to propose CQ/ HCQ treatment and the inflammatory reaction caused by SARSCoV-2 infection. Given its location and function in the body, it acts as a producer of ROS, which participate in the processes of sepsis and respiratory distress. In this sense, many works have attempted to test antioxidant therapy as co adjuvants in the treatment of these pathological processes. However QR2 as a specific target has been studied for cancer related processes and not for these other pathologies [39,40].
The selection of drugs with potential to inhibit QR2 was made based on the characteristics of the enzyme and its most common inhibitors. The planarity of the molecule is a constant feature in inhibitors of enzyme families of Phase II drug metabolism. The cyclic conformation of the compounds is another trait to consider, and as reported in other studies, the para-amino group in the transstilbene benzene ring is essential for aromatase inhibitory activity, and the introduction of an imidazole moiety improves activity greatly. Primary and tertiary amines, chloroquine among them, are protonated; however, it seems more relevant to determine the presence of heterocyclic nitrogen and its relative location in the identification of new compounds. On the other hand, QR2 uses a unique catalytic site for its substrate and co-substrate, this determines that the rigidity of their chemical structure and their steric hindrance, determines an accentuated effect on their behavior, determining a preference for molecules with multiple conjugates double bonds such as catecholamines quinones [19,41]. Another peculiarity of the compounds that interact with QR2 is that they can do in different oxidation states, and that the binding affinity varies depending on whether the enzyme is in the oxidized or reduced state. Analyzing a differentiation between QR2 inhibitors for different oxidation states could have an impact at the physiological level [19]. This is why features such as Number of hydrogen bond donors and Number of hydrogen bond acceptors were included for the constitution of the data set. The selection of characteristics, although it was carried out under the processes described in the body of the text, was based on the knowledge of the particularities described above, which helped the analysis by groups which is presented below.
In Group I, we consider the relevance of the compounds of the camostat and report for being bioassays against SARS-CoV-2; and 5-Aminopyrimidine due to its relationship with QR2. In COVID-19, the camostat has been shown to potently inhibit the serine protease TMPRSS2, necessary for the interaction between the viral S glycoprotein and the ACE2 receptor, preventing its entry into the cell [42]. Its use could be doubly beneficial in SARS-CoV-2 infection since, by unknown mechanisms, it suppresses the pathways associated with oxidative stress [43]. Relacatib inhibits the activity of cathepsin K, a cysteine protease that breaks down type I collagen involved in osteoporosis, osteoarthritis, and other disorders that cause bone breakdown. Its use in COVID-19 is tested because it was shown to also inhibit cathepsin L, critical for the entry of the virus into the cell [44]. The nucleus aminopyrimidine can be found in various biologically active compounds. Inhibition of QR2 by compounds with an aminopyrimidine ring has been demonstrated. For example, the drugs quercetin, imatinib and nilotinib, used to treat leukemia, have an aminopyrimidine nucleus and inhibit QR2 [45]. The action of compounds with aminopyrimidine in their structure has also been associated with anti-inflammatory action [46].Roscovitine, a 2,6,O9-trisubstituted purine used as an antineoplastic, strongly reduces acute lung inflammation secondary to bacterial infection [47]. However, the inhibition of QR2 by these compounds and their association with oxidative status is not clear, in some cases they suppress oxidative stress [48] and in others they induce it [49]. With respect to 5-Aminopyrimidine, several derivatives have demonstrated their antioxidant capacity by modifying cellular metabolism [50]. Due to its antioxidant action and detection by the algorithm, we believe that camostat, relacatib, 5-Aminopyrimidine and its derivatives could have an inhibitory effect on QR2 activity.
Because the binding of dopamine to QR2 is well known [51], we decided, in Group II, to screen for drugs with a chemical structure similar to dopa. It stands out, among 23 compounds identified as Active, N-Caffeoyl DOPA or clovamide, an amide isostere of rosmarinic acid. Clovamide andseveral of its analogues have shown anti-inflammatory action [52]. Park et al., propose the use of some of them as anti-inflammatory agents by demonstrating that they suppress the overproduction of nitric oxide in microglial cells [53]. The antioxidant action of clovamide analogs has been demonstrated in other works, as well as its antinitrative effect [54,55]. Therefore, we believe that the actions of clovamide derivatives on QR2 should be investigated.
For its benzoquinone ring, we chose Group III screening for substances derived from ubiquinone (coenzyme Q10), a natural compound widely distributed in the body. In its reduced form, ubiquinone is a powerful lipophilicantioxidant[56]. It has a side chain composed of 10 isoprene units attached to the benzoquinone ring. Ubiquinone can be artificially synthesized which helps to treat various diseases [57]. Several derivatives were found by the model as Active, among them Coenzyme Q4 (CoQ4), classified with a threshold of 0.99997, the best in the group. Although short chain quinones are toxic, especially those with 0 to 3 units of isoprene, CoQ4 with 4 units show minimal toxicity [58]. Another quinone with an isoprene chain greater than 3, found as Active, was Decylubiquinone (Dub) a member of the 1,4-benzoquinone class which is 2,3-dimethoxybenzoquinone which has been substituted at positions 5 and 6 by decyl and methyl groups. For its classification threshold 0.99743 and have shown that Dub inhibits the production of ROS [59,60] could be with CoQ4 a good candidate as a substitute drug CQ/HCQ.
Among the Group IV bioassay drugs, the human monoclonal antibody sarilumab, used for the treatment of Rheumatoid Arthritis (RA), is was tested in patients with severe and critical COVID-19 [61]. In RA, sarilumab was associated with decreased acute phase inflammatory biomarkers [62]. The second of the compounds that stands out for its threshold is fingolimod, an aminodiol consisting of propane-1,3-diol, modulator of the Sphingosine 1-Phosphate Receptor (SP) used for the treatment of multiple sclerosis. Of the 5 isoforms of SP1 (S1P1-5): S1P1; S1P2; and S1P3 are expressed ubiquitously, while the expression of S1P4 and S1P5 receptors does so in the nervous and immune systems [63]. Fingolimod has been shown to decrease ROS production in mitochondria by restoring function and morphology. Although some of its effects are due to interaction with the SP1 receptor, the mechanisms of action of some of its effects are not well known [64]. Finally, rivaroxaban, an oxazolidinone derivative, is used as an oral anticoagulant since it inhibits the factor Xa. There is evidence that factor Xa has proinflammatory effects, therefore [65]. However, it has recently been shown to have protective effects against oxidative stress and mitochondrial toxicity [66]. Due to their chemical structure, sarilumab and fingolimod could have effects similar to CQ/HCQ in SARS-CoV-2. In the case of rivaroxaban, although identified by the model as Active, its use in COVID-19 would be inadvisable due to the risk of bleeding complications, especially if it is combined with antiviral drugs since they raise their plasma concentration [67].
In Group V, amantadine, a member of the adamantane class, is used as an antiviral, antiparkinsonian, and pain reliever. Amantadine, identified by the algorithm as Active against QR2 with a threshold of 0.99152, is an N-Methyl-D-Aspartate (NMDA) receptor antagonist whose antioxidant and anti-inflammatory effects have been demonstrated in nervous tissue [68]. Recently Tipton and Wszolek, they suggested the potential benefit of amantadine in SARS-CoV-2 infection, since it has been shown that bananin, a derivative thereof, inhibits viral replication of other coronaviruses [69]. Pramipexole, in Group V as well, is a member of the class of benzothiazoles. Pramipexole is a selective dopamine receptor agonist used in Parkinson’s disease with demonstrated mitochondrial and neuroprotective antioxidant action [70]. Obtained a high rating threshold.
B vitamins, screened in Group VI, act as antioxidants by several well-known mechanisms [71]. Of the group, the compounds prosultiamine and alinamin, derived from vitamin B1, were identified as Active. The first, prosultiamine, is converted to vitamin B1 after intestinal absorption, and is successfully used in vitamin B1 deficiency diseases such as beriberi and Wernicke’s encephalopathy [72]. Although vitamin B1 was not identified as Active, the finding of prosultiamine and alinamin as possible QR2 inhibitors opens the possibility of finding vitamin B1 derivatives as QR2 inhibitors in the future.
In Group VI, 11 vitamins of the K complex were identified. The interaction between vitamins K and quinones reductases is not clear. For example, it is believed that the reaction between QR2 and vitamin K3 could produce cytotoxicity [73,74]. Vitamins of the K complex could bind QR2 and inhibit it, however, its use in COVID-19 could be limited by thrombotic complications [75].
Conclusions
It was presented a series of compounds identified by the QSAR model as QR2 inhibitors and we analyze the main drugs in that series according to their availability and current use. The main idea was to demonstrate how, from the application of ML techniques, compounds that were already available could be identified, making it possible to test them faster in the treatment of COVID-19, given that the expansion rate of the disease and its consequences requires the use of strategies that reduce the selection times of therapeutic approaches, always supported by the available evidence.
References
- Wu Z, McGoogan JM. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA-Journal of the American Medical Association. American Medical Association. 2020; 323: 1239-1242.
- Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak-An update on the status. Military Medical Research. BioMed Central Ltd. 2020: 7.
- Schoeman D, Fielding BC. Coronavirus envelope protein: Current knowledge. Virology Journal. BioMed Central Ltd. 2019: 16.
- Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on DecadeLong Structural Studies of SARS Coronavirus. J Virol. 2020; 94.
- Song W, Gui M, Wang X, Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018; 14.
- Abassi ZA, Skorecki K, Heyman SN, Kinaneh S, Armaly Z. Covid-19 infection and mortality - A physiologist’s perspective enlightening clinical features and plausible interventional strategies. American journal of physiology. Lung cellular and molecular physiology. NLM (Medline). 2020.
- Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005; 2.
- Marella A, Tanwar OP, Saha R, Ali MR, Srivastava S, Akhter M, et al. Quinoline: A versatile heterocyclic. Saudi Pharmaceutical Journal. 2013; 21: 1-12.
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research. Springer Nature; 2020; 30: 269-271.
- Gao J, Tian Z, Yang X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. BioScience Trends. International Advancement Center for Medicine and Health Research Co. Ltd. 2020: 14.
- Molina JM, Delaugerre C, Le Goff J, Mela-Lima B, Ponscarme D, Goldwirt L, et al. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Medecine et Maladies Infectieuses. Elsevier Masson SAS. 2020.
- Haeusler IL, Chan XHS, Guérin PJ, White NJ. The arrhythmogenic cardiotoxicity of the quinoline and structurally related antimalarial drugs: A systematic review. BMC Med. 2018; 16.
- Juurlink DN. Safety considerations with chloroquine, hydroxychloroquine and azithromycin in the management of SARS-CoV-2 infection. CMAJ. 2020; 192: E450-E453.
- Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARSCoV-2 infection in vitro. Cell Discovery. Springer Nature; 2020: 6.
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosomelysosome fusion. Autophagy. 2018;14: 1435-1455.
- Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol. 2002; 62(1): 364-372.
- Kwiek JJ, Haystead TAJ, Rudolph J. Kinetic Mechanism of Quinone Oxidoreductase 2 and Its Inhibition by the Antimalarial Quinolines. Biochemistry. 2004;43: 4538-4547.
- Human NAD(P)H:quinone oxidoreductase2. Gene structure, activity, and tissue-specific expression. PubMed-NCBI. 2020. Available from: https://www.ncbi.nlm.nih.gov/pubmed/?term=H uman+NAD+(P)+H%3A+quinone+oxidoreductase2.+Gene+structu re%2C+activity%2C+and+tissue-specific+expression
- Buryanovskyy L, Fu Y, Boyd M, Ma Y, Hsieh TC, Wu JM, et al. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry. 2004;43: 11417-11426.
- Jockers R, Maurice P, Boutin JA, Delagrange P. Melatonin receptors, heterodimerization, signal transduction and binding sites: What’s new? British Journal of Pharmacology. 2008; 54: 1182-1195.
- Gould NL, Elkobi A, Edry E, Daume J, Rosenblum K. MuscarinicDependent miR-182 and QR2 Expression Regulation in the Anterior Insula Enables Novel Taste Learning. eneuro. 2020.
- Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Seminars in Immunopathology. Springer Verlag. 2017; 39: 529-539.
- Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The experience of clinical immunologists from China. Clinical Immunology. Academic Press Inc. 2020; 214.
- Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J Crit Care. 2020.
- Multicenter collaboration group of Department of Science and Technology of Guangdong Province and Health Commission of Guangdong Province for chloroquine in the treatment of novel coronavirus pneumonia. [Expert consensus on chloroquine phosphate for the treatment of novel coronavirus pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43: 185-188. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32164085
- Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov EN, Andrade CH. QSAR-based virtual screening: Advances and applications in drug discovery. Frontiers in Pharmacology. Frontiers Media S.A. 2018; 9.
- Lin X, Li X, Lin X. A review on applications of computational methods in drug screening and design. Molecules. 2020; 25.
- Knox RJ, Jenkins TC, Hobbs SM, Chen S, Melton RG, Burke PJ. Bioactivation of 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB 1954) by human NAD(P)H quinone oxidoreductase 2: A novel cosubstrate-mediated antitumor prodrug therapy. Cancer Res. 2000; 60: 4179-4186.
- Chan K, Leung HCM, Tsoi JKH. Predictive QSAR model confirms flavonoids in Chinese medicine can activate voltage-gated calcium (CaV) channel in osteogenesis. Chinese Med (United Kingdom). 2020; 15.
- Maiti A, Reddy PVN, Sturdy M, Marler L, Pegan SD, Mesecar AD, et al. Synthesis of casimiroin and optimization of its quinone reductase 2 and aromatase inhibitory activities. J Med Chem. 2009; 52: 1873-1884.
- Reddy PVN, Jensen KC, Mesecar AD, Fanwick PE, Cushman M. Design, synthesis, and biological evaluation of potent quinoline and pyrroloquinoline ammosamide analogues as inhibitors of quinone reductase 2. J Med Chem. 2012; 55: 367-377.
- John SE, Jensen KC, Kang S, Chen Y, Calamini B, Mesecar AD, et al. Design, synthesis, biological and structural evaluation of functionalized resveratrol analogues as inhibitors of quinone reductase 2. Bioorganic Med Chem. 2013; 21: 6022-6037.
- Yang JH, Kondratyuk TP, Jermihov KC, Marler LE, Qiu X, Choi Y, et al. Bioactive compounds from the fern Lepisorus contortus. J Nat Prod. 2011; 74: 129-136.
- Leclerc V, Ettaoussi M, Rami M, Farce A, Boutin JA, Delagrange P, et al. Design and synthesis of naphthalenic derivatives as new ligands at the melatonin binding site MT3. Eur J Med Chem. 2011; 46: 1622-1629.
- Conda-Sheridan M, Marler L, Park EJ, Kondratyuk TP, Jermihov K, Mesecar AD, et al. Potential chemopreventive agents based on the structure of the lead compound 2-bromo-1-hydroxyphenazine, isolated from streptomyces species, strain CNS284. J Med Chem. 2010; 53: 8688-8699.
- Du H, Wang J, Zhang X, Hu Z. A novel quantitative structure-activity relationship method to predict the affinities of MT3 melatonin binding site. Eur J Med Chem. 2008; 43: 2861-2869.
- Tropsha A. Best practices for QSAR model development, validation, and exploitation. Vol. 29, Molecular Informatics. 2010; 476-488.
- Gómez-Jiménez G, Gonzalez-Ponce K, Castillo-Pazos DJ, Madariaga-Mazon A, Barroso-Flores J, Cortes-Guzman F, et al. The OECD Principles for (Q)SAR Models in the Context of Knowledge Discovery in Databases (KDD). In: Advances in Protein Chemistry and Structural Biology. Academic Press Inc. 2018; 85-117.
- Reybier K, Perio P, Ferry G, Bouajila J, Delagrange P, Boutin JA, et al. Insights into the redox cycle of human quinone reductase 2. Free Radic Res. 2011; 45: 1184-1195.
- Vella F, Ferry G, Delagrange P, Boutin JA. NRH:quinone reductase 2: An enzyme of surprises and mysteries. Biochemical Pharmacology. Elsevier Inc. 2005; 71: 1-12.
- Foster CE, Bianchet MA, Talalay P, Zhao Q, Amzel LM. Crystal structure of human quinone reductase type 2, a metalloflavoprotein. Biochemistry. 1999; 38: 9881-9886.
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020; 181: 271-280.e8.
- Shuto T, Kamei S, Nohara H, Fujikawa H, Tasaki Y, Sugahara T, et al. Pharmacological and genetic reappraisals of protease and oxidative stress pathways in a mouse model of obstructive lung diseases. Sci Rep. 2016; 6.
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune crossreactivity with SARS-CoV. Nat Commun. 2020; 11.
- Winger JA, Hantschel O, Superti-Furga G, Kuriyan J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct Biol. 2009; 9: 7.
- Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, et al. Roscovitine in cancer and other diseases. Annals of Translational Medicine. AME Publishing Company. 2015; 3.
- Hoogendijk AJ, Roelofs JJTH, Duitman JW, van Lieshout MHP, Blok DC, van der Poll T, et al. R-roscovitine reduces lung inflammation induced by Lipoteichoic acid and Streptococcus pneumoniae. Mol Med. 2012; 18: 1086-1095.
- Liu L, Zhou J, Wang Y, Qi T, Wang Z, Chen L, et al. Imatinib inhibits oxidative stress response in spinal cord injury rats by activating Nrf2/HO‑1 signaling pathway. Exp Ther Med. 2020; 19: 597-602.
- Gajski G, Gerić M, Domijan AM, Golubović I, Garaj-Vrhovac V. Evaluation of oxidative stress responses in human circulating blood cells after imatinib mesylate treatment – Implications to its mechanism of action. Saudi Pharm J. 2019; 27: 1216-1221.
- Procházková E, Jansa P, Dracinsky M, Holy A, Mertlikova-Kaiserova H. Determination of the antioxidative activity of substituted 5-aminopyrimidines. Free Radic Res. 2012; 46: 61-67.
- Fu Y, Buryanovskyy L, Zhang Z. Quinone reductase 2 is a catechol quinone reductase. J Biol Chem. 2008; 283: 23829-23835.
- Lim HW, Park JI, More SV, Park JY, Kim BW, Jeon SB, et al. Antineuroinflammatory effects of DPTP, a novel synthetic clovamide derivative in in vitro and in vivo model of neuroinflammation. Brain Res Bull. 2015;112: 25-34.
- Park JY, Kim BW, Lee HU, Choi DK, Yoon SH. Synthesis of clovamide analogues that inhibit no production in activated BV-2 microglial cells. Biol Pharm Bull. 2017;40: 1475-1482.
- Kolodziejczyk-Czepas J, Krzyżanowska-Kowalczyk J, Sieradzka M, Nowak P, Stochmal A. Clovamide and clovamide-rich extracts of three Trifolium species as antioxidants and moderate antiplatelet agents in vitro. Phytochemistry. 2017;143: 54-63.
- Hu XL, Lin J, Lv XY, Feng JH, Zhang XQ, Wang H, et al. Synthesis and biological evaluation of clovamide analogues as potent antineuroinflammatory agents in vitro and in vivo. Eur J Med Chem. 2018;151: 261-271.
- Raizner AE. Coenzyme Q10. Methodist DeBakey cardiovascular journal. NLM (Medline); 2019; 5: 185-191.
- Shukla S, Dubey KK. CoQ10 a super-vitamin: review on application and biosynthesis. 3 Biotech. Springer Verlag. 2018; 8.
- Cerqua C, Casarin A, Pierrel F, Vazquez Fonseca L, Viola G, Salviati L, et al. Vitamin K2 cannot substitute Coenzyme Q 10 as electron carrier in the mitochondrial respiratory chain of mammalian cells. Sci Rep. 2019; 9.
- Devun F, Walter L, Belliere J, Cottet-Rousselle C, Leverve X, Fontaine E. Ubiquinone analogs: A mitochondrial permeability transition pore-dependent pathway to selective cell death. PLoS One. 2010; 5: 1-8.
- Cao J, Liu X, Yang Y, Wei B, Li Q, Mao G, et al. Decylubiquinone suppresses breast cancer growth and metastasis by inhibiting angiogenesis via the ROS/p53/ BAI1 signaling pathway. Angiogenesis. Angiogenesis. 2020.
- Evaluation of the Efficacy and Safety of Sarilumab in Hospitalized Patients With COVID-19 - Full Text View-ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT04315298
- Gabay C, Burmester GR, Strand V, Msihid J, Zilberstein M, Kimura T, et al. Sarilumab and adalimumab differential effects on bone remodelling and cardiovascular risk biomarkers, and predictions of treatment outcomes. Arthritis Res Ther. 2020; 22.
- Ahmed N, Linardi D, Muhammad N, Chiamulera C, Fumagalli G, Biagio LS, et al. Sphingosine 1-phosphate receptor modulator fingolimod (FTY720) attenuates myocardial fibrosis in postheterotopic heart transplantation. Front Pharmacol. 2017; 8.
- Martín-Montañez E, Pavia J, Valverde N, Boraldi F, Lara E, Oliver B, et al. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic Biol Med. 2019; 137: 116-130.
- Kirchhof P, Ezekowitz MD, Purmah Y, Schiffer S, Meng IL, Camm AJ, et al. Effects of Rivaroxaban on Biomarkers of Coagulation and Inflammation: A Post Hoc Analysis of the X-VeRT Trial. TH Open. 2020;04: e20-e32.
- Samiei F, Sajjadi H, Jamshidzadeh A, Seydi E, Pourahmad J. Contrasting Role of Concentration in Rivaroxaban Induced Toxicity and Oxidative Stress in Isolated Kidney Mitochondria. Drug Res (Stuttg). 2019;69: 523-527.
- Testa S, Prandoni P, Paoletti O, Morandini R, Tala M, Dellanoce C, et al. Direct oral anticoagulant plasma levels striking increase in severe COVID‐19 respiratory syndrome patients treated with antiviral agents. The Cremona experience. J Thromb Haemost. 2020.
- Dogan G, Karaca O. N-methyl-D-aspartate Receptor Antagonists may Ameliorate Spinal Cord Injury by Inhibiting Oxidative Stress: An Experimental Study in Rats. Turk Neurosurg. 2020;30(1):60–8.
- Tipton PW, Wszolek ZK. What can Parkinson’s disease teach us about COVID-19? Neurol Neurochir Pol. 2020; 54: 204-206.
- Spiess AN, Neumeyer N. An evaluation of R2as an inadequate measure for nonlinear models in pharmacological and biochemical research: A Monte Carlo approach. BMC Pharmacol. 2010; 10.
- Moretti R, Peinkhofer C. B vitamins and fatty acids: What do they share with small vessel disease-related dementia?. International Journal of Molecular Sciences. 2019; 20.
- Therapeutic benefits of an oral vitamin B1 derivative for human T lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis (HAM - PubMed - NCBI. 2020. Available from: https://www.ncbi.nlm.nih.gov/pubmed/?term=Therapeutic+be nefits+of+an+oral+vitamin+B1+derivative+for+human+T+lymph otropic+virus+type+I-associated+myelopathy%2Ftropical+spasti c+paraparesis+(HAM%2FTSP)
- Gong X, Gutala R, Jaiswal AK. Quinone Oxidoreductases and Vitamin K Metabolism. Vitamins and Hormones. 2008; 78: 85- 101.
- NQO2 inhibition relieves reactive oxygen species effects on mouse oocyte meiotic maturation and embryo development.-PubMedNCBI. Available from: https://www.ncbi.nlm.nih.gov/bmed/?term =NQO2+inhibition+relieves+reactive+oxygen+species+effects+on +mouse+oocyte+meiotic+maturation+and+embryo+development
- Vivas D, Roldán V, Roldán R, Asuncí On Esteve-Pastor M, Roldán I, Tello A, et al. Journal Pre-proof Recomendaciones sobre el tratamiento antitrombóticoantitromb´antitrombótico durante la pandemia COVID-19. Posicionamiento del Grupo de Trabajo de Trombosis Cardiovascular de la Sociedad Espã nola de Cardiología. Rev Española Cardiol.2020. Available from: https:// doi.org/10.1016/j.recesp.2020.04.006