
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Primary leiomyosarcoma of femur: A rare entitycase report and review of literature
Nouha Ben Abdeljelil; Asma Ben Mabrouk*; Ahlem Bellalah; Manel Njima; Rim Hadhri; Abdelfattah Zakhama
Department of Pathological Anatomy and Cytology, Fattouma Bourguiba University Hospital, Monastir, Tunisia.
*Corresponding Author : Asma Ben Mabrouk
Department of Pathological Anatomy and Cytology,
Fattouma Bourguiba University Hospital, Monastir,
Tunisia.
Email: Asma.benmabrouksaid@yahoo.com
Received : Mar 05, 2021
Accepted : Apr 16, 2021
Published : Apr 20, 2021
Archived : www.jcimcr.org
Copyright : © Mabrouk AB (2021).
Abstract
Background: We report this case to highlight clinical and pathological features of this rare entity.
Methods: It is about a 48 year-old woman with an osteolytic lesion of the lower extremity of the left femur.
Result: A biopsy of the lesion showed densely cellular malignant mesenchymal proliferation. It was made by bundles of smooth muscle cells with nuclei of moderate to severe atypia. The immunohistochemical study showed an intense and diffuse cytoplasmic positivity of tumor cells with the smooth anti-muscle. Desmin was focal positive. After neoadjuvant chemotherapie, a knee block resection was made. Macroscopically, it was a 10 cm sized tumor located in the distal part of tumor. Histologically tumor showed post chemotherapy effects estimated at 20%. The bone and muscle surgical limits were unscathed.
Conclusion: Primary leiomyosarcoma of bone is a rare and a diagnostically challenging tumor entity.
Keywords: Leimyosarcoma; Bone; Surgery; Chemotherapy
Abreviations: POL: Primary Osseous Eiomyosarcoma; HPF: High-Power Field; UPS: Undifferentiated Pleomorphic Sarcoma; P63: Protein63.
Citation: Abdeljelil NB, Mabrouk AB, Bellalah A, Njima M, Hadhri R, et al. Primary leiomyosarcoma of femur: A rare entityCase report and review of literature. J Clin Images Med Case Rep. 2021; 2(2): 1054.
Introduction
Primary Osseous Leiomyosarcoma (POL) is a very rare sarcoma, accounting for <0.7% of all primary malignant bone tumors [1]. It was firstly reported by Evans and Sanerkin in 1965 [2]. Since then, case reports along with few case series have been published on this relatively uncommon entity. POL shows histologic, immunohistochemical, and ultrastructural features of smooth muscle differentiation. It is a high-grade destructive tumor and has a poor prognosis with limited treatment options. Herein, we report this case to highlight clinical and pathological features of this rare entity.
Case report
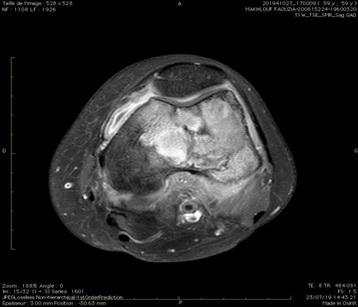
The patient was a 48 year-old woman presented a painful tumefaction of the left thigh evolving since one year and increased in size. Imaging showed an epiphyso-metaphyso- diaphyseal osteolytic lesion of the lower extremity of the left femur (Figure 1).
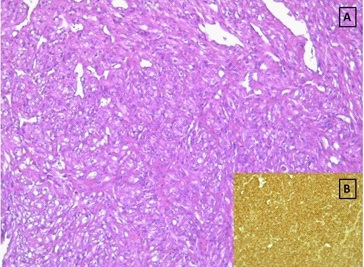
A biopsy of the lesion showed densely cellular malignant mesenchymal proliferation focally necrotic. It was made by bundles of smooth muscle cells with hyperchromatic nuclei of moderate to severe atypia with fairly numerous figures of mitosis (Figure 2A). The tumor contains numerous vessels. The immunohistochemical study showed an intense and diffuse cytoplasmic positivity of tumor cells with the Smooth Muscle Actin (SMA) (Figure 2B). Desmin was focal positive. Epithelial markers (Cytokeratin, EMA) and vascular markers (CD31, CD34) were negative. Based on the histological and immunochemical features of the tumor, the diagnosis of POL was made.
The patient received four courses of neo-adjuvant chemotherapy and had a knee block resection. Macroscopically the tumor measured 10 cm in size and was located in the distal part of the femur. It had a beige color, a firm consistency and fasciculated appearance (Figure 3).

Microscopic examination revealed that the tumor showed fibrous end edematous reshuffles. This appearance was due to post chemotherapy effects estimated at 20% of the tumor surface. The tumor infiltrated focally the cortical without exceeding it. The bone and muscle surgical limits were unscathed.
Within the follow-up of 6 months, no clinical symptoms and signs of tumor recurrence were detected in our case and her clinical outcome was good.
Discussion
POL is a rare entity that affects mainly adults. Males and females are almost equally affected [3]. The median patient age is 47 years, but cases affecting a wide age range (9-88 years) have been reported [3]. The long bones are the most affected site, mainly at the knee region involving proximal tibia and distal femur [4,5] (as in this case). Most of POL arising in long bones involves the metaphysis with or without an extension to the epiphysis, diaphysis, or surrounding soft tissue [6]. Other sites of involvement in descending order of frequency include craniofacial skeleton, pelvic bones, proximal humerus, clavicle and vertebrae [3,5,6]. POL may arise de novo or in association with prior radiation(15% of cases) [5]. Immunocompromised patients may develop smooth muscle tumors associated with EBV invariable organs, which are designated as EBV-associated smooth muscle tumors and may rarely involve the bone [6-8]. Pain and swelling are the most frequent complaint and other presentations include a mass and/or pathological fracture. On imaging, generally, there is a low index of suspicion for diagnosis of a primary intraosseous soft tissue sarcoma, including POL. The principal radiological feature consists in a solitary osteolytic radiolucent intramedullary mass with indistinct margins and cortical destruction with no presence of bone matrix production [1,3]. Histopathologic analysis represents the gold standard for the diagnosis of POL. Gross examination shows an intramedullary bone tumor with a grey-tan or creamy white color, fleshy or fibrous texture, cortical destruction, extension into soft tissue, and frequent pathologic fracture. The size of the tumor ranges from 2 to 12 cm with an average of 6.1 cm [3,5,9]. The histopathologic characteristics of primary bone LMS are identical to those arising from other more common anatomic sites, showing the same morphological and phenotypic features as smoothmuscle differentiation. The classic morphological pattern of POL is represented by spindle cells, which are usually disposed in fascicles and intersect at perpendicular angles. Tumor cells show cytological atypia with abundant, deeply eosinophilic, fibrillar cytoplasm and elongated, cigar shaped nuclei with occasional subnuclear vacuoles [1,3,10]. In a series of 33 cases of POL, Antonescu et al. tried to classify leiomyosarcomas of the bone into high grade (26 cases) and low grade tumors (7 cases) by an assessment of their histologic parameters, including degree of cellularity, cellular pleomorphism or anaplasia, mitotic activity degree of necrosis, and invasive growth [9].
Lower-grade sarcomas are typically cellular and consist of spindle cells with uniform, blunt ended nuclei with cigar-shaped morphology. Necrosis and brisk mitotic activity are usually absent. Higher-grade sarcomas demonstrate overt nuclear pleomorphism, hyperchromasia, and prominent nucleoli. As in any high-grade sarcoma, mitotic figures and tumor necrosis are common features [3].
POL diagnosis is characterized by the absence of either osteoid or chondroid matrix [1]. Osteoclast-likegiant cells may be present in variable quantities. Other secondary elements such as prominent, ectatic staghorn vessels have been reported [9]. Some tumors have extensive extracellular stromalmatrix, which may consist of homogenized, hyalinizing fibrosis or myxohyaline matrix or thick, wirelike collagen bundles [3,11,12]. This classical type is the most common end follwed by epithelioid or clearcell, myxoid and pleomorphic variants [9].
Epithelioid leiomyosarcoma is a recognized variant that has been described in the uterus and soft tissue as well as bone [3,13,14]. As its name suggests, the tumor cells are large and rounded. The cytoplasmic borders are well defined, and the cytoplasm is abundant and eosinophilic. The central nuclei often have vesicular chromatin with conspicuous nucleoli.
Occasionally, vacuolization may be especially prominent and encompass nearly the entire cytoplasm, imparting a clear cell morphology [3].
Immunohistochemical stains are helpful, especially in poorly differentiated tumors. Similar to its uterine and soft tissue counterparts, tumor cells in POL are consistently uniformly positive for both Smooth Muscle Actin (SMA) and Muscle-Specific Actin (MSA) in more than 95% and 93% of cases respectively [3,10,15] However, desmin is positive in only 50% of cases and thus should not be used as the only primary screening antibody [3,5,10,15]. It should be noted that cytokeratin and S100 protein expression have been reported. Cytokeratin can be focally positive in more than 30% of cases, especially in the epithelioid variant [3,10]. S100 protein was positive in 4 of 5 cases in one study, and 1 of 20 cases in another [16,17]. Together, absent desmin expression and positive cytokeratin or S100 protein expression are confounding factors that contribute to the difficulty in diagnosing POL.
POL presents a differential diagnosis problem when it is low grade with leiomyoma and due to the rarity of this two entities, the criteria for their differentiation are not well defined. Nuclear atypia and pleomorphism, mitotic activity and necrosis have been used in the differentiation. Mitotic activity is the best marker for malignancy. A benign leiomyoma should have a mitotic count of no greater than 4/50 HPF [5].
The main differential diagnoses of high grade POL include metastatic leiomyosarcoma particularly from the female genital tract and gastrointestinal tract, fibrosarcoma, primary Undifferentiated Pleomorphic Sarcoma (UPS), osteosarcoma, and metastatic sarcomatoid carcinoma [3,5]. Metastatic leiomyosarcoma can be reliably diagnosed only by clinical history and must be excluded before a diagnosis of POL of bone can be rendered [3]. If there is involvement of surrounding soft tissues, this raises the possibility of a primary soft tissue leiomyosarcoma secondarily involving bone. It has been suggested that if the epicentre is primarily within soft tissue and/or the bulk of tumour is within soft tissue as determined radiologically or by gross examination, then the tumour should be considered a soft tissue primary [9]. Although there is no consensus on the proportion of intraosse ous tumour required to label a tumour as arising within bone, a cut-off of greater than 70% has been proposed [9]. POL and fibrosarcoma have the same clinical and radiographic findings. Morphologically, both entities are composed of spindle cells in an orderly fascicular pattern. However fibrosarcoma classically demonstrates a herringbone pattern and the cells have tapered rather than blunt ended nuclei and usually more intercellular collagen [5]. Diffuse and intense positivity for muscle markers is in favor of POL. Myofibroblastic differentiation may be found in fibrosarcoma, and although this is generally focal, it may still lead to a mistaken diagnosis of leiomyosarcoma [4]. UPS shows greater pleomorphism, lacks a prominent interlacing fascicular pattern and blunt ended nuclei. It is commonly a cellular proliferation of spindle and epithelioid cells with highgrade and bizarre cytology and frequent mitoses and necrosis. In some cases, focal myxoid stroma, brisk inflammation, and/ or giant cells may be prominent features [18]. However, UPS displays ambiguous histomorphologic and immunohistochemical features, and is therefore usually a diagnosis of exclusion [18,19]. The judicious use of immunohistochemical markers such as SMA and desmin is especially helpful in distinguishing UPS from POL [3]. Lack of osteoid or chondroid matrix and negativity of muscle markers excludes osteosarcoma. However, a recent study showed that 20% of POL within its cohort contained non neoplastic or dystrophic calcification, a finding that may be mistaken for true malignant osteoid/bone formation [20]. Metastatic sarcomatoid carcinoma may morphologically mimic POL. As mentioned above, POL can had a focal positivity for cytokeratin so that immunohistochemistry for epithelial markers: Broad-spectrum cytokeratinor EMA, lack of myogenic markers, and presence of other specific markers such as P63 in squamous cell carcinoma and PAX-8 in renal cell carcinoma, as well as clinical history are effective strategies to discern between metastatic sarcomatoid carcinoma and POL.
The treatment of choice for POL is wide surgical resection with clear margins, and the role of chemotherapy and radiotherapy has not yet been established because of the lack of uniformity in treatment and the limited number of cases reported. In a study of 33 patients, Antonescu et al [9] did not find any difference in survival rates between patients who received surgery alone and patients who received surgery and adjuvant radiation therapy. Although the tumor stage and histologic grade have been described to be correlated with overall and disease-free survivals in POL. For example, Antonescu et al [9] reported in their series that 5- year survival rates in patients with high-grade tumors is 60% versus 100% in low-grade. And in a review study by Adelani et al [10], the 5-year overall survival rates in patients with stage I and IIA tumors were 90% and 60%, respectively. In contrast, the numbers were reduced to 29% and 0% in patients with stage IIB and III/IV tumors, respectively. Other prognosis factors, found in many studies, are described: Negative surgical margins and the absence of metastasis at the time of diagnosis which both have been linked to better survival outcomes [15], unlike the radiation- associated POL which has been linked to worse survival outcomes [9]. The rate of local recurrence appears to be similar between high-grade and low- grade tumors with 29% and 33% for high grade and low grade tumors respectively [9,15]. The metastatic rate at 5 years was higher in highgrade tumors (58%) than low-grade tumors (33%) [9]. Highlighting the aggressive behavior of POL, in a study by Rekhi et al [20], all metastatic cases appeared within the first year of diagnosis. However, metastatic disease may develop after a long time interval from the initial therapy, particularly in low grade tumors [10]. Lung is the most common metastatic site, followed by axial skeleton and liver.
Conclusion
POL is a rare primary sarcoma of bone. An index of suspicion for its diagnosis is necessary in cases of osteolytic, destructive lesions, without matrix production. Histopathologic evaluation with immunohistochemical analysis is helpful in exact recognition. It may mimic a number of other primary sarcomas of bone; in particular fibrosarcoma and UPS. Treatment is primarily surgical with limited benefit from neoadjuvant and adjuvant therapies. Prognosis appears to be dismal with metastasis occurring primarily to the lung.
References
- Recine F, Bongiovanni A, Casadei R, Pieri F, Riva N, De Vita A, et al. Primary leiomyosarcoma of the bone: A case report and a review of the literature. Medicine (Baltimore). 2017; 96: e8545.
- Evans DM, Sanerkin NG. Primary leiomyosarcoma of bone. J Pathol Bacteriol. 1965; 90: 348-350.
- Wang GY, Lucas DR. Primary Leiomyosarcoma of Bone: Review and Update. Arch Pathol Lab Med. 2019; 143: 1332‑1337.
- Amstalden EMI, Barbosa CSP, Gamba R. Primary leiomyosarcoma of bone: Report of two cases in extragnathic bones. Annals of Diagnostic Pathology. 1998; 2: 103‑110.
- Khor TS, Sinniah R. Leiomyosarcoma of the bone: A case report of a rare tumour and problems involved in diagnosis. Pathology. 2010; 42: 87‑91.
- Matsuyama A, Sakamomo A, Aoki T, Hisaoka M. Intraosseous leiomyosarcoma arising in the epiphysis of the distal femur. Pathol Res Pract. 2013; 209: 530‑533.
- To KF, Lai FM, Wang AY, Leung CB, Choi PC, Szeto CC, et al. Posttransplant Epstein- Barr virus-associated myogenic tumors involving bone. Cancer. 2000; 89: 467‑472.
- Deyrup AT, Lee VK, Hill CE, Cheuk W, Toh HC, et al. Epstein-Barr virus- associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: a clinicopathologic and molecular analysis of 29 tumors from 19 patients. Am J Surg Pathol. 2006; 30: 75‑82.
- Antonescu CR, Erlandson RA, Huvos AG. Primary leiomyosarcoma of bone: A clinicopathologic, immunohistochemical, and ultrastructural study of 33 patients and a literature review. Am J Surg Pathol. 1997; 21: 1281‑1294.
- Adelani MA, Schultenover SJ, Holt GE, Cates JMM. Primary leiomyosarcoma of extragnathic bone: Clinicopathologic features and reevaluation of prognosis. Arch Pathol Lab Med. 2009; 133: 1448‑1456.
- Chow LTC. Metatarsal leiomyosarcoma masquerading as acute osteomyelitis - A diagnostic trap unveiled by vigilant clinical, radiologic and pathologic analysis. Foot (Edinb). 2016; 27: 10-5.
- Berlin O, Angervall L, Kindblom LG, Berlin IC, Stener B. Primary leiomyosarcoma of bone. A clinical, radiographic, pathologic-anatomic, and prognostic study of 16 cases. Skeletal Radiol. 1987; 16: 364‑376.
- Yamamoto T, Minami R, Ohbayashi C, Inaba M. Epithelioid Leiomyosarcoma of the External Deep Soft Tissue. Archives of Pathology & Laboratory Medicine. 2002; 126: 468‑470.
- Cui RR, Wright JD, Hou JY. Uterine leiomyosarcoma: A review of recent advances in molecular biology, clinical management and outcome. BJOG. 2017; 124: 1028‑1037.
- Mori T, Nakayama R, Endo M, Hiraga H, Tomita M, et al. Fortyeight cases of leiomyosarcoma of bone in Japan: A multicenter study from the Japanese musculoskeletal oncology group. J Surg Oncol. 2016; 114: 495‑500.
- Wirbel RJ, Verelst S, Hanselmann R, Remberger K, Kubale R, Mutschler WE. Primary leiomyosarcoma of bone: clinicopathologic, immunohistochemical, and molecular biologic aspects. Ann Surg Oncol. 1998; 5: 635‑641.
- Khoddami M, Bedard YC, Bell RS, Kandel RA. Primary leiomyosarcoma of bone: report of seven cases and review of the literature. Arch Pathol Lab Med. 1996; 120: 671‑675.
- Matushansky I, Charytonowicz E, Mills J, Siddiqi S, Hricik T, Cordon-Cardo C. MFH classification: Differentiating undifferentiated pleomorphic sarcoma in the 21st Century. Expert Rev Anticancer Ther. 2009; 9: 1135‑1144.
- Goldblum JR. An approach to pleomorphic sarcomas: can we subclassify, and does it matter? Mod Pathol. 2014; 27: S39-46.
- Rekhi B, Kaur A, Puri A, Desai S, Jambhekar NA. Primary leiomyosarcoma of bone--a clinicopathologic study of 8 uncommon cases with immunohistochemical analysis and clinical outcomes. Ann Diagn Pathol. 2011; 15: 147‑156.