
Journal of Clinical Images and Medical Case Reports
ISSN 2766-7820
Case Report - Open Access, Volume 2
Palisaded neutrophilic and granulomatous dermatitis, A critical diagnosis in a severe lupus flare
Stephanie L Kuschel; Isabelle M Sanchez; Michelle Bain; Maria Tsoukas*
University of Illinois at Chicago, Department of Dermatology, Chicago, IL, USA.
*Corresponding Author : Maria Tsoukas
University of Illinois Hospital & Health Sciences
System, 808 S Wood St, Chicago, IL 60612, USA
Email: tsoukasm@uic.edu & skuschel@uic.edu
Received : Apr 01, 2021
Accepted : Apr 28, 2021
Published : Apr 30, 2021
Archived : www.jcimcr.org
Copyright : © Tsoukas M (2021).
Abstract
Palisaded Neutrophilic And Granulomatous Dermatitis (PNGD) is a rare inflammatory cutaneous eruption associated with underlying systemic disease, commonly Systemic Lupus Erythematosus (SLE), rheumatoid arthritis, or Anti-Neutrophilic Cytoplasmic Autoantibody (ANCA)-associated vasculitis. Appropriate diagnosis is challenging, requiring coordination of care between medicine, dermatology, pathology, and rheumatology.
We describe a 29-year-old female with SLE who developed eroded papules and erythematous, edematous plaques on the ear anti-helices, hands, and elbows during hospitalization for workup of acute on chronic abdominal pain. Biopsies showed an interstitial infiltrate of zonal collagen necrobiosis, consistent with typical PNGD histopathology, involving collagen inflammation surrounded by palisading neutrophils, histiocytes, karyorrhexis, and leukocytoclastic vasculitis.
The association of ANCA-vasculitis makes differentiation between underlying autoinflammatory disease in PNGD from new vasculitis difficult; however, histopathology is key to distinguishing them. Treatment should focus on improvement of the underlying autoimmune condition. Adjuvant treatments are intralesional or systemic corticosteroids, hydroxychloroquine, or dapsone. Given the complexities of associated conditions, it is important to bring awareness of PNGD to the medical community for improved diagnostic accuracy and subsequent care.
Keywords: Complex medical dermatology; Inpatient dermatology; Palisaded neutrophilic and granulomatous dermatitis; Palisaded neutrophilic granulomatous dermatitis; Systemic lupus erythematosus.
Abbreviations: SLE: Systemic lupus erythematosus; ANCA: antineutrophil cytoplasmic antibodies; PNGD: Palisaded neutrophilic and granulomatous dermatitis; IVIG: Intravenous immune globulin.
Citation: Kuschel SL, Sanchez IM, Bain M, Tsoukas M. Palisaded neutrophilic and granulomatous dermatitis, A critical diagnosis in a severe lupus flare. J Clin Images Med Case Rep. 2021; 2(2): 1096.
Introduction
Palisaded and Neutrophilic Granulomatous Dermatitis (PNGD) is a rare cutaneous manifestation of systemic illness, most commonly an auto-immune condition. Immune-complex deposition is believed to initiate an inflammatory cascade which results in the development of papules, with varying forms of secondary morphology (crusting, erosions, umbilications, etc.), primarily on the dorsal fingers, elbows, and ear anti-helices. Due to its rarity, PNGD may be underdiagnosed or misdiagnosed; however, if recognized, it can aid in the diagnosis of an underlying systemic illness. In patients with a known history of auto-immune conditions, the development of PNGD has been associated with an increased incidence of lupus nephritis and a tendency to demonstrate a good response to systemic steroids [1]. Regardless of the timing of its onset, management of the underlying condition remains the mainstay of care for PNGD.
Case presentation
Dermatology was consulted for evaluation of pruritic, tender, edematous, red papules on the upper extremities and ears of a 29-year-old female with a history of Systemic Lupus Erythematosus (SLE), Charcot Marie Tooth disease, and gastroparesis. She was hospitalized for acute on chronic abdominal pain, nausea, and vomiting. Her home medications included mycophenolate mofetil 500 mg twice daily and prednisone 5 mg daily.
Examination revealed several eroded papules on the ear anti-helices, many erythematous papules on the elbows, and several larger, targetoid, violaceous papules with a blanching halo on the dorsal fingers (Figure 1).
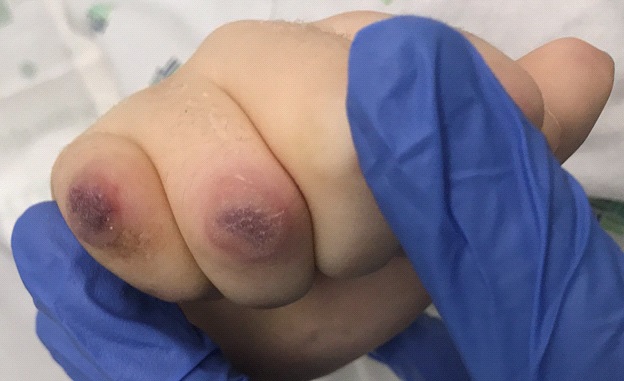
Laboratory data showed transaminitis and an elevated antidouble-stranded DNA titer. Initially, complement levels were normal, but later decreased precipitously. Antineutrophil Cytoplasmic Antibodies (ANCA) specific for myeloperoxidase were positive.
Punch biopsies revealed a dense, diffuse, interstitial infiltrate forming a zonal pattern of collagen necrobiosis accompanied by an overlying, epidermal blister due to marked spongiosis (Figure 2). Granular IgM deposition was seen along the dermalepidermal junction, confirming a diagnosis of Palisaded Neutrophilic and Granulomatous Dermatitis (PNGD).
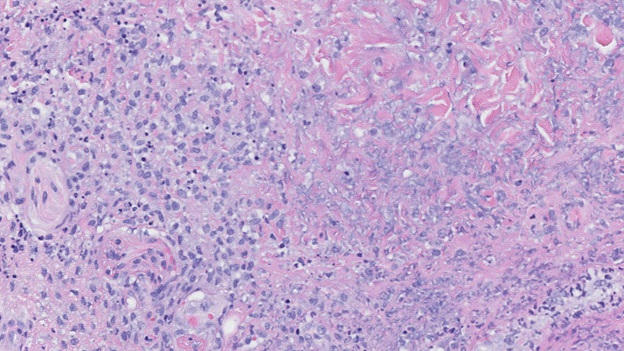
The patient’s home doses of Mycophenolate and prednisone were increased respectively to 1 g twice daily and 20 mg daily. The patient transitioned to methylprednisolone 30 mg/kg/day when her clinical course worsened. Intravenous Immune Globulin (IVIG) 2 g/kg was given and plans were made to start rituximab or cyclophosphamide.
Unfortunately, the patient’s hospital course was complicated by her development of respiratory failure requiring mechanical ventilation. She developed seizures secondary to central nervous system vasculitis, and her overall clinical decline was concerning for a severe lupus flare with multi-organ involvement. She failed to improve despite high-dose corticosteroids, and her family elected for palliative extubation one month after her initial presentation.
Discussion
PNGD is a rare cutaneous eruption associated with an underlying auto-immune disease (commonly SLE, rheumatoid arthritis, or an ANCA-associated vasculitis), or malignancy. Up to 31% of systemic lupus patients also have positive ANCAs, especially when vascular disease is considered, however, its importance in the pathogenesis of SLE is unresolved [2]. Nevertheless, ANCA vasculitides or an SLE-ANCA overlap syndrome are relevant considerations [2]. In patients with a known history of SLE, the development of PNGD has been associated with an increased incidence of lupus nephritis and a tendency to demonstrate a good response to systemic steroids; however, relapsing skin disease has been observed with attempted tapering of therapy [1]. In our case, the patient developed signs of renal compromise during the course of her hospitalization. Plans for a renal biopsy to evaluate for lupus nephritis were deferred due to the patient becoming systemically unstable. A renal biopsy is helpful to differentiate lupus nephritis from an ANCA-associated glomerulonephritis. However, further studies are needed to determine whether ANCAs in patients with lupus nephritis are associated with more severe renal involvement [3].
PNGD is clinically manifested by skin-colored to erythematous, edematous papules. These papules may appear umbilicated, eroded, or crusted, and present symmetrically on extensor surfaces, typically on the elbows and dorsal fingers [4].
The pathogenesis of PNGD arises from immune complexes causing perivascular damage and inducing a small vessel neutrophilic vasculitis, which gradually contributes to poor perfusion to the dermis. The collagen degenerates and stimulates the palisading lympho-histiocytic infiltrate [5]. Histopathology of early lesions shows a dermal infiltrate of neutrophils. Leukocytoclastic vasculitis and karyorrhexis (nuclear fragmentation and debris) may also be present. Late lesions are characterized by zones of collagen breakdown surrounded by palisading histiocytes, neutrophils, or nuclear debris [6].
Treatment of PNGD primarily relies on adequate treatment of the underlying disease. There are no prospective trials regarding therapies; however, cases in the literature report improvement with intralesional or systemic corticosteroids, hydroxychloroquine, or dapsone. Topical corticosteroids are not effective [7].
Diagnosis of PNGD is challenging, requiring multidisciplinary care. Given the complexities of associated conditions, it is important to bring awareness of PNGD to the medical community for improved diagnostic accuracy and subsequent care.
References
- Terai S, Ueda-Hayakawa I, Nguyen CTH, Ly NTM, et al. Palisaded neutrophilic and granulomatous dermatitis associated with systemic lupus erythematosus: Possible involvement of CD163 + M2 macrophages in two cases, and a review of published works. Lupus. 2018; 27: 2220-2227.
- Itikyala S, Pattanaik D, Raza S. Systemic Lupus Erythematosus (SLE) and antineutrophil cytoplasmic antibody-associated vasculitis (AAV) overlap syndrome: Case report and review of the literature. Case Rep Rheumatol. 2019; 2019: 5013904.
- Turner-Stokes T, Wilson HR, Morreale M, Nunes A, Cairns T, et al. Positive antineutrophil cytoplasmic antibody serology in patients with lupus nephritis is associated with distinct histopathologic features on renal biopsy. Kidney Int. 2017; 92: 1223-1231.
- Rosenbach MA, English JC. Reactive granulomatous dermatitis: A review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015; 33: 373-387.
- Rosenbach MA, Wanat KA, Risenauer A, et al. Non-infectious granulomas. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Philadelphia: Elsevier Saunders. 2018: 1659- 1661.
- Ko CJ, Glusac E. Lever’s Histopathology of the Skin, 11th ed.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad of Dermatol. 2011; 65: e92-93.